

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
The NCBI Handbook [Internet]. 2nd edition. Bethesda (MD): National Center for Biotechnology Information (US); 2013-.
This publication is provided for historical reference only and the information may be out of date.

Scope
The Database of Genotypes and Phenotypes (dbGaP) is a National Institutes of Health (NIH) sponsored repository charged to archive, curate and distribute information produced by studies investigating the interaction of genotype and phenotype (1). It was launched in response to the development of NIH’s GWAS policy and provides unprecedented access to very large genetic and phenotypic datasets funded by National Institutes of Health and other agencies worldwide. Scientists from the global research community may access all public data and apply for controlled access data.
The information contained in dbGaP includes individual level molecular and phenotype data, analysis results, medical images, general information about the study, and documents that contextualize phenotypic variables, such as research protocols and questionnaires. Submitted data undergoes quality control and curation by dbGaP staff before being released to the public.
Information about submitted studies, summary level data, and documents related to studies can be accessed freely on the dbGaP website (http://www.ncbi.nlm.nih.gov/gap). Individual-level data can be accessed only after a Controlled Access application, stating research objectives and demonstrating the ability to adequately protect the data, has been approved (https://dbgap.ncbi.nlm.nih.gov/aa/wga.cgi?page=login). Public summary data from dbGaP are also accessed without restriction via the PheGenI tool, as detailed in the Related tools section.
History
Planning for the database began in 2006 and the database received its first request for data in mid-2007. The initial release of dbGaP contained data on two Genome-Wide Association Studies (GWAS): the Age-Related Eye Diseases Study (AREDS), a 600-subject, multicenter, case-controlled, prospective study of the clinical course of age-related macular degeneration and age-related cataracts supported by the National Eye Institute (NEI), and the National Institute of Neurological Disorders and Stroke (NINDS) Parkinsonism Study, a case-controlled study that gathered DNA, cell line samples and detailed phenotypic data on 2,573 subjects. The data from the Genetic Association Information Network (GAIN) (2) was released soon after.
Although initially designed for GWAS, the scope of dbGaP has expanded to facilitate making individual level information accessible to research communities and to provide data needed to understand the manifestation of disease and how that relates to the genome, proteome and epigenome. The dbGaP has been growing rapidly since its inception. See the dbGaP home page for current content.
Data Model
Accessioned Objects
The data in dbGaP are organized as a hierarchical structure of studies. Accessioned objects within dbGaP include studies, phenotypes (as variables and datasets), various molecular assay data (SNP and Expression Array, Sequence, and Epigenomic marks), analyses, and documents (Figure 1). Each of these is described in its own section below.

Figure 1.
This figure shows the relationships between dbGaP accessioned objects and whether they are available publicly or only through Controlled Access. This is an updated version of a figure that originally appeared in Mailman, et al. 2007.
Studies
The data archived and distributed by dbGaP are organized as studies. Studies may be either stand-alone or combined in a “parent study/child study” hierarchy. Parent or “top level” studies may have any number of child studies (also referred to as substudies). However, study hierarchy is limited to two levels (parent and child only). In other words, substudies may not have substudies. Studies, whether parent or child, can contain all types of data ascertained in genetic, clinical or epidemiological research projects such as phenotype and molecular assay information that are linked via subject and sample IDs. Studies often contain documents, such as questionnaires and protocols, which help contextualize the phenotype and genotype data. Study data are distributed by consent groups, each of which contains all data from a set of study participants who have signed the same consent agreement. In other words, the data delivered for a single consent group will all have the same Data Use Limitations (DULs) for future research use.
Each study is assigned a unique accession number which should be used when citing the study. The general dbGaP accession format for a study is phs######.v#.p#. The first three letters [phs] denote the object type (‘s’ denotes study), followed by 6-digit, 0-padded object number (######) which is consecutively assigned by dbGaP. The version number (.v#) indicates updates of the object, where # is initially 1 and increments by 1 as the object is updated. The version number is followed by participant group (.p#) where # is initially 1 and increments by 1 as the participant group changes. The version number of a study will increment any time the version of an object contained by the study (such as a phenotype variable, genotype data, or a sub study) is updated. The participant group of a study will change when existing subjects are removed, or when an existing subject changes from one consent group to another, but not when additional subjects are added.
While the data found in studies can vary widely based on both the number of participants and the variety of deposited phenotypic and molecular data, all studies include basic descriptive metadata such as study title, study description, inclusion/exclusion criteria, study history, disease terms, publications related to the study, names and affiliations of the principal investigators, and sources of funding. This information is publicly available on the study’s report page at the dbGaP website.
Datasets and Variables
Phenotypic data values are submitted to dbGaP as tabular files or datasets (accessioned with pht#, where ‘t’ denotes table), where columns represent phenotypic variables (accessioned with a phv#, where ‘v’ denotes variable) and rows represent subjects. A dbGaP phenotype variable consists of two parts: the data values and the description of the data in the accompanying data dictionary. Each cell (value) in a dataset is stored in a relational database and is mapped to the appropriate phenotype variable and subject. Phenotype variable metadata are provided by the submitter via a data dictionary for each dataset and include: variable name, variable description, units, and a list of any coded responses. The variable’s data type (text string, integer, decimal or date) is automatically determined by calculating which type is in the majority. Conflicts between submitted and calculated data types, or other discrepancies in the data, are reconciled by dbGaP curators in consultation with the data submitter.
Variables are created from the columns of the dataset; each variable and dataset is accessioned using the general dbGaP format ph(v|t)######.v#.p#. A variable’s version (v#) will change when either values of data change or its entry in the data dictionary changes. A dataset’s version will change when a variable inside the dataset is added, updated or deleted. For both variables and datasets the participant set (p#), is inherited from the study to which it belongs. Variables, and sometimes datasets, are linked to appropriate sections of documents (please see the Website section below for details).
Individual level phenotype data is only available through the dbGaP Authorized Access System. Public summary-level variable information is available on the dbGaP website and ftp site.
Genotype data
Genotype data hosted at the dbGaP consist of individual level genotypes and aggregated summaries, both of which are distributed through the dbGaP Authorized Access System. The types of data available include DNA variations, SNP assay, DNA methylation (epigenomics), copy number variation, as well as genomic/exomic sequencing. RNA data types such as expression array, RNA seq, and eQTL results are also available. For details about the accepted format of submitted genotype files please see the dbGaP submission guide.
Genotype data are accessioned based on their data type and use the general dbGaP accession format ph(g|e|a)######.v# where ‘g’ denotes GWAS, ‘e’ expression, and ‘a’ analysis. Versioning of genotype data is triggered by addition or withdrawal of samples, sample consent status change, or error correction.
Genotype data files are compressed and archived into tar files for distribution. The files are explicitly named to indicate file content, such as image data (cel and idat), genotype calls (genotype), and locus annotations (marker info). Genotype calls are usually clustered according to file format and genotyping platform, including one sample per file (indfmt), multiple-sample matrix (matrixfmt) and pre-defined variant call format (vcf). They will be accompanied by sample-info file for subject lookup and consent status. The consent code and consent abbreviation are also embedded in the file name.
Examples of genotype data file names:
phe000005.v1.FHS_SHARe_project4_miRNA.sample-info.MULTI.tar
phg000006.v6.FHS_SHARe_Affy500K.genotype-calls-matrixfmt.c2.HMB-NPU-MDS-IRB.tar
The various pieces of the names can be parsed to extract meaningful content: the genotype accession (phe000005.v1 and phg000006.v6); the study (FHS_SHARe in both cases); the molecule type (miRNA); the platform/chip information (Affy500K); the content type (sample-info or genotype-calls-matrixfmt); the consent code (c2); and the consent abbreviation (HMB-NPU-MDS-IRB).
Analyses
Because of the large volume of data generated and concerns regarding participant confidentiality many genetic epidemiological analyses have not been published. But, because individual-level data is only accessed through Controlled Access, dbGaP can archive, integrate and distribute these results.
Analyses can either be provided by submitters or be pre-computed by dbGaP staff, though pre-computes account for a small number of the total analyses. Submitted analysis results are accessioned with the prefix “pha”. After removing identifiable elements, like counts and frequencies, analysis results are displayed in the public dbGaP browser that dynamically links to NCBI annotation resources, like dbSNP, Gene, RefSeq. These public views can be found through the “Analysis” link on the study page and they can be downloaded from the FTP site. The original submitted analyses, including updated marker info, are fully accessible through dbGaP Controlled Access.
Analysis files are typically formatted by population, trait and analysis method (typical), however some include surveys across multiple populations, and may use either SNPs or genes as the loci analyzed. However, in general, they all contain the following three parts which are also required for dbGaP submission.
- 1.
Metadata, which includes trait, population, sample size and brief descriptions on Analysis and Method.
- 2.
Marker information and genotyping summary, such as identifiers of loci (variation, gene and structure variant), alleles, genotype counts (frequency), call rate and p-value from Hardy-Weinberg-equilibrium testing.
- 3.
Testing statistics, including p-value, effect size (odds ratio/regression coefficient/relative risk) and direction (coding allele) if association results.
With these resources, other scientists can verify the discoveries, recalculate statistics under various genetic models, develop new hypotheses, and more importantly, construct a meta-analysis even though individual–level data are inaccessible. The details of data fields are listed in dbGaP submission guides and we welcome suggestions and comments from the scientific community. An interactive view of the analsysis results submitted to dbGaP is described in The dbGaP Genome Browser in the Related Tools section of this chapter.
Documents
The dbGaP encourages investigators to submit documents related to their studies, such as protocols, patient questionnaires, survey instruments and consent forms, along with their data. These documents provide valuable information and context for subsequent researchers who will apply for and download datasets. All submitted documents are available publicly and can be used by anyone interested in gaining a better understanding of the phenotypic data found in a study.
Each document is accessioned using the general dbGaP format phd######.v# where “d” indicates document. A document’s version (v#) will change when the variables annotated on the document change, or when the document itself is changed significantly. (For example, fixing a typo would not be considered a significant change unless it were to change the meaning of the document.)
Documents submitted to dbGaP are represented in a common XML format. Converting documents into a common format allows all documents to be treated uniformly in the database (aiding indexing and discovery) and to be displayed in a single HTML style. Additionally, the XML format allows curated information to be added to the documents. This curated information is used to create live links between the documents and other portions of the dbGaP website, such as variable report pages. Linking between documents and other objects will be discussed further in the section about the dbGaP website.
Documents are generally viewable on the dbGaP website in both HTML and PDF format (the PDF for a document may be the originally submitted object, if it was sent as PDF, or could be a PDF representation of another format such as Microsoft Word or Excel or a plain text file).
The XML used by dbGaP is an extension of NLM’s Archiving and Interchange Tag Set Version 2.3 (http://dtd.nlm.nih.gov/archiving/2.3/). The extension adds structures to code questionnaires and adds a number dbGaP-specific attributes to common document structures (such as sections, tables, and lists) to facilitate curation. A copy of our extension is publicly available at http://dtd.nlm.nih.gov/gap/2.0/wga-study2.dtd, and documentation for the extension is located at http://dtd.nlm.nih.gov/gap/2.0/doc/wga-document.html.
Dataflow
Submissions
The NIH strongly supports the broad sharing of de-identified data generated by NIH-funded investigators and facilitates data sharing for meritorious studies that are not NIH-funded. Decisions about whether non-NIH-funded data should be accepted are made by individual NIH Institutes and Centers (IC); ICs will not accept data unless the submission is compatible with NIH’s GWAS policy.
NIH-Funded Studies
Institutional certification, as well as basic information about the study, is required when submitting data to dbGaP.
- Institutional certification consists of a letter signed by the principal investigator and an institutional official that confirms permission to submit data to dbGaP. NIH has developed Points to Consider for IRBs and Institutions to assist institutions in their review and certification of an investigator’s plan for submission of data to dbGaP.
- Basic information consists of items like the title of the study, a description and history of the study, inclusion and exclusion criteria, listing of preview and certification of PI’s data
- rincipal investigators (PIs) and funding information.
Principal investigators (PIs) should familiarize themselves with the “NIH Points to Consider” document that provides information about: the NIH GWAS Data Sharing Policy; benefits of broad sharing of data through a central data repository; risks associated with the submission and subsequent sharing of such data; safeguards designed to protect the confidentiality of research participants; and specific points for institutional review boards (IRBs) to consider during review and certification of PIs’ data submission plans.
The principal investigators must contact their NIH program official (PO) to begin the submission process. If the study was not funded by the NIH, PIs should contact vog.hin.mln.ibcn@pleh-PaGbd for guidance.
Non-NIH-Funded Studies
To submit non-NIH-funded data to dbGaP the following information will need to be provided:
- Institutional certification as described in the last section. To provide this, someone from the institution or organization will need to be registered in eRA Commons. Information regarding registration is available from the eRA Commons website. (Note: The review of a PI’s request can be initiated without the certification, but the review process will be expedited if GWAS staff receives the certification at time of submission.)
- Basic information about the study, as described in the previous section.
- The NIH IC that most closely aligns with the research. A list of ICs can be found at http://www.nih.gov/icd/.
- Whether the study has been published or accepted for publication. If it has the PI should provide documentation (i.e., the publication citation or a copy of any correspondence indicating that an article about the study has been accepted for publication).
The PI should submit all information and the certification to vog.hin.liam@SAWG. Once GWAS staff receives the documents, they will forward them to the appropriate IC program administrator for consideration. The IC program administrator will contact the PI with any questions and/or to notify you of the IC’s decision.
The PI is encouraged to consult with the Program Officer/Director (PO/PD) and/or IC GWAS Program Administrator (GPA) at an NIH Institute or Center (IC) to discuss the project, data sharing plan, and data certification process (non NIH funded projects should contact pleH PaGbd) to complete the registration process.
Instructions for submitters
Study Registration
Before data can be submitted to dbGaP, the study must be registered in the dbGaP Registration system following these steps:
- The GPA from the sponsoring IC gathers study registration information from the PI.
- Completes the study registration in the dbGaP Registration System by providing:
- Study details
- Signed Institutional Certification
- Approved Data Use Certification (DUC)
- Consent groups and Data Use Limitations (DUL)
- The Registration System sends an automated email to the investigator upon completion of the study registration acknowledging the study registration and giving further instructions on how to submit data to dbGaP.
Data submission
The PI will be provided with the dbGaP Submission Guide. The packet contains templates and instructions on how to format the data files for submission to dbGaP. The expected files for each single study are:
- 1_dbGaP_StudyConfig*
- 2a_dbGaP_SubjectPhenotypesDS
- 2b_dbGaP_SubjectPhenotypesDD
- 3a_dbGaP_SampleAttributesDS*
- 3b_dbGaP_SampleAttributesDD*
- 4a_dbGaP_SubjectDS*
- 4b_dbGaP_SubjectDD*
- 5a_dbGaP_SubjectSampleMappingDS*
- 5b_dbGaP_SubjectSampleMappingDD*
- 6a_dbGaP_PedigreeDS**
- 6b_dbGaP_PedigreeDD**
- *
Required
** Required if there are related subjects
Note for studies that expect/involve SRA (Sequence Read Archive) file submission
Once the required files (listed above) are received by dbGaP, passed dbGaP QCs, and the subject consents and subject sample mapping have been loaded into dbGaP and provided to BioSample, the PI will be provided with a study accession number and a link to the corresponding study sample status page. The SRA submitter can then apply for an SRA submission account (Aspera account) and submit SRA files to dbGaP.
Data processing
Data received at dbGaP undergoes a sequence of processing steps. Figure 2 illustrates the steps involved in moving a study through dbGaP. The first step is getting the study registered (as has been discussed above), and occurs before data transmission (shown in gray). The dbGaP data processing occurs in two pipelines, the phenotype curation (blue) and genotype curation (purple). These pipelines are largely processed in parallel but converge prior to data release. The final step is preparing the study for release to the public (green). Particulars of the phenotype and genotype curation will be discussed below.
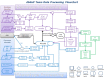
Figure 2.
This figure shows the complex processing that occurs after data has been submitted to dbGaP. Of particular note is the step, in the center of the chart in black, where samples are matched.
Phenotype processing
The phenotypic data are subjected to both automated and human-mediated assessment.
Before the data are loaded into the database, scripts are used to evaluate the following:
- Poor formatting - Each dataset submitted to dbGaP should be a rectangular table showing variables in columns and subject or sample IDs in rows.
- HIPAA violations - The datasets submitted to dbGaP should follow the Health Insurance Portability and Accountability Act (HIPAA) rules in order to protect the privacy of personally identifiable health information.
- Issues with Subject and Sample IDs - Submitters are required to use the subject consent file to list all the subjects who participated in, or are referred to by, the study, with their consent values. Subjects who had not directly participated in the study, e.g., parents of participants included in the pedigree file, should also be included in the consent file with consent value 0.
- Missing information in Data Dictionaries - The submitters are required to submit data dictionaries, in addition to datasets, to explain the meanings of the variables and data values. For each value in the datasets, dbGaP requires that the submitters provide a variable description, variable type, units of values for numerical variables, and logical minimum and maximum values if available. For each encoded value, a code meaning should be included in the data dictionary.
- Issues with Pedigree files- A pedigree file submitted to dbGaP should include the following columns: family ID, subject ID, father ID, mother ID, sex, and twin ID if available. We also require that all the subjects who appear in father ID or mother ID columns also be included in the subject ID column.
More detailed information about the particular automated testing performed to catch errors in the broad categories listed above can be found in Appendix – Phenotype Quality Control.
Reports from the automated quality control (QC) scripts are reviewed by curatorial staff. If necessary, curators will communicate with the submitters and ask that new files be submitted correcting the errors. Even if the automated QC checks do not detect problems, the curatorial staff check all data dictionaries manually to see if there are any problems that were not identified by automated checks.
Genotype processing
The genotype data processing and QC process consists of the following steps:
- 1.
Check for availability of Sample-Subject Mapping file and validate against subject list.
- 2.
Check for availability of sample genotype file and validate against SSM.
- 3.
- 4.
Check for data consistency in the submitted QC component against data from other submissions for the study.
- 5.
Conduct QC checks and generate genotype-QC component. This step includes checking:
- a.
Missing call rates per sample/per marker.
- b.
Minor allele frequency.
- c.
Mendelian error rate when trios are available.
- d.
Duplicate concordance check (Generate SNP and subject filters based on tests results
- e.
when dups are available).
- f.
Gender check.
- g.
IBD analysis.
- 6.
Generate SNP and subject filters based on tests results.
- 7.
Verify genotype and phenotype data using NCBI GWAS pre-compute against similar pre-compute provided by PI/analysis group.
- 8.
Split PLINK sets according to consent and generate genotype-calls-mtrxfm components.
- 9.
Generate sample-info and marker-info release components.
- 10.
Partition and pack build of individual genotype files according to subject consent information and data type.
The quality of the genotype data is checked at both the genotype data file and the genotyping level. Typically at the file level a genotype release contains individual level data in both individual (one file per sample) and matrix (one matrix with all samples) formats. The genotype matrices are generated by dbGaP curators from submitted individual genotype files and subject related information, such as gender and pedigree data. These matrices then are used to generate pre-computes/metrics/QC-filters, which are further verified against similar pre-computes submitted by the investigators. When necessary, the genotyping quality of each sample is also verified using a B-Allele frequency (BAF) analysis pipeline which calculates and processes BAF values to identify samples with extremely “noisy” or failed genotyping.
The following quality assurance steps are implemented to facilitate cross-study and cross-technology data merging and analysis:
- 1.
Identify duplicated genotypes across all studies as well as generating data.
- 2.
Check data formats, annotation, and QC- metrics for genotype data derived using different technologies.
- 3.
Check ID reconciliation, gender, missing call rate, duplicate concordance, identity-by-descent, and Mendelian error rate at sample as well as SNP level.
Subjects and Samples
In dbGaP there are two similar yet distinct concepts that describe the participants in a study: subject and sample. A subject corresponds to an individual human. A sample in dbGaP corresponds to each analyte (DNA/RNA) that is put in the machine or on the chip, rather than the physical result of obtaining a tissue or blood sample, though this is accepted as well. Modeling samples this way allows dbGaP to track duplicates, centers, plates, wells, and any sequence of aliquots that precedes the actual aliquot used to produce the molecular data finally submitted to dbGaP. The tracking method also enables dbGaP to easily add, rename, or redact samples over time.
Example
Consider a case in which a single subject has both a blood draw and a cheek swab. DNA is extracted from both samples. The DNA extracted from the blood is stored for years after being drawn, and then sequenced on two different platforms, and the DNA extracted from the cheek swab is also used on a GWAS chip. In this scenario, dbGaP prefers to receive three samples belonging to the same subject (even though there were two intermediate physical samples).
Note: The information about intermediate samples may be informative and can be included as one or more variables in the sample attribute file.
Submitters are required to assign de-identified IDs to subjects and samples; these are submitted subject and sample ids. However, dbGaP also assigns a unique id to samples and subjects; these are the dbGaP Subject and Sample IDs and are included in the final phenotype files available through controlled access. The dbGaP assigns its own IDs to accurately represent cases where a single subject (person) has participated in more than one study. In such a case the two submitted subjects will be assigned the same dbGaP Subject ID. This can only be done if the submitter provides the information that a subject in their study is the same subject as in an existing dbGaP study. Similarly, cell repository, or otherwise readily available samples such as Coriell samples, used as controls in multiple studies will typically receive the same dbGaP Sample ID.
All phenotype and molecular data are connected though the Subject Sample Mapping file.
Data ID mapping
The data submitted to the dbGaP are de-identified. The phenotype and genotype data are connected through the subject sample mapping file in which one sample is mapped to exactly one subject and one subject is mapped to any number of samples. The following is a partial list of the IDs and attributes included in dbGaP phenotype and molecular data files.
- 1.
SUBJECT_ID: This is the submitted Subject ID and it is included in the Subject Consent Data File, the Subject Sample Mapping Data File, the Pedigree Data File (if applicable), and all Subject Phenotype Data Files. SUBJECT_ID should be an integer or string value consisting of the following characters: English letters, Arabic numerals, period (.), hyphen (-), underscore (_), at symbol (@), and the pound sign (#). In addition to the submitted Subject ID, dbGaP will assign a dbGaP Subject ID that will be included in the final phenotype dump files along with the submitted Subject ID.
- 2.
SAMPLE_ID: This is the submitted Sample ID and is included in the Subject Sample Mapping Data File and the Sample Attributes Data File. This ID should be used as the key for the individual level molecular data. Each sample should be submitted with a single, unique, de-identified Sample ID. The acceptable characters in Sample IDs are the same as those in the Subject IDs. In addition to the submitted Sample ID, dbGaP will assign a dbGaP Sample ID that will be included in the final phenotype dump files along with the submitted Sample ID. The SAMPLE_IDs listed in the Subject Sample Mapping Data File should be identical to the samples found in the genotype, SRA, and other molecular data.
- 3.
dbGaP_SAMPLE_ID: This is the dbGaP assigned unique identifier assigned to the submitted Sample ID. The dbGaP Sample ID is included as a column in the final phenotype dump files whenever there is a submitted sample ID column.
- 4.
dbGaP_SUBJECT_ID: This is the dbGaP unique identifier assigned to the submitted Subject ID. The dbGaP Subject ID is included as a column in the final phenotype dump files whenever there is a submitted subject ID column. The dbGaP Subject ID is unique cross all dbGaP studies, which means that if a subject is known to have participated in multiple studies that have been submitted to dbGaP, the same dbGaP Subject ID will be assigned to the individual across multiple studies, though the submitted subject ID may be different.
- 5.
SOURCE_SUBJECT_ID and SUBJECT_SOURCE: The Source Subject ID (SOURCE_SUBJECT_ID) is the de-identified alias Subject ID used in the public repository, consortium, institute, or study from where the subject has been obtained. The Subject Source (SUBJECT_SOURCE) is the name of the third party source, public repository, consortium, institute, or study that corresponds to the subject. For subjects originating from a shared source (such as a public repository, consortium, institute, study, etc.) or for subjects with alias IDs, these 2 variables will be included in the Subject Consent Data File. The SOURCE_SUBJECT_ID maps to the SUBJECT_ID. For referencing HapMap subjects from Coriell, the SUBJECT_SOURCE value is written as “Coriell.” The SOURCE_SUBJECT_ID should be written as the de-identified subject ID assigned by Coriell (e.g., NA12711).
- 6.
FAMILY_ID: The Family ID is a column of de-identified Family IDs in the pedigree file. The Family ID is also referred to as the Pedigree ID. The family ID should be the same for individuals belonging in the same biological family. The family ID is found in the pedigree file if a pedigree file is available.
- 7.
SEX: The gender variable can be included in a subject phenotype data file or in a pedigree file if a pedigree file is available.
- 8.
FATHER and MOTHER: In the pedigree file, FATHER and MOTHER are the two columns of the unique, de-identified subject IDs of the participant’s biological father and mother. The Father ID and Mother ID may not be identical. 0 (zero) or blank is filled in for founders or marry-ins (parents not specified) in a pedigree. Each unique Father ID and unique Mother ID is also listed in the Subject ID column of both the Pedigree Data File and the Subject Consent Data File.
- 9.
TWIN_ID: Monozygotic twins or multiples of the same family have Twin IDs. Twins or multiples of the same family share the same TWINID, but are assigned different SUBJECT_IDs.
- 10.
CONSENT: Every subject that appears in a Subject Phenotype Data File must belong to a single consent group and every sample that appears in a Subject Sample Mapping File and in a Sample Attribute Data File must belong to a consented subject. The consent information is listed in the Subject Consent Data File. Consents are determined by the submitter, their IRB, and their GPA (GWAS Program Administrator) along with the DAC (Data Access Committee). All data is parsed into its respective consent groups for download.
Curatorial document annotation
One thing that sets dbGaP apart from similar databases is the extent of curatorial work done with the data and documentation we receive. For documents, this involves making connections between appropriate portions of text and other accessioned objects (such as variables, data tables, and other documents) and creating links to external resources. We refer to this process as “document annotation” and it involves embedding references into the XML for the documents.
Documents can either be annotated by the submitter or by the dbGaP curator responsible for a study. Types of annotations include adding variable IDs to particular sections of text so that the text can be linked to the variable report page or adding references so that hyperlinks can be made between chapters of a protocol document.
Access
Public data (unrestricted)
dbGaP Website
Report types
The web site provides reports specific to the objects in dbGaP. These reports are explained in the following.
Study
- the study’s accession, name, description, history, inclusion/exclusion criteria, a summary of the molecular data collected, a list of related publications, and a list of relevant phenotypes selected by the PI;
- links to Authorized Access, description of data use limitations and use restrictions, release date, embargo release date, and a list of users and their public and technical research use statements who have been authorized to access individual-level data;
- links to publicly available information – including a study manifest -- via a public ftp site;
Variable
- the variable’s name, accession, description, comments;
- a statistical summary of the variable’s values;
- a curated list of excerpts from study documents that relate to the variable
Document
- the document’s name and accession;
- the document’s contents in HTML format; note that the red question marks link particular excerpts of the document to other study objects. For example, clicking on a red question mark near a protocol description might list the phenotype variables that were measured using that protocol;
- a link to a PDF version of the document
Analysis
- the analysis’ name, accession, description, and a brief synopsis of the methods used;
- relevant summary plots (e.g. Manhattan plots of p-values; Log QQ p-value plot);
- a link to the Genome Browser, where analysis results can be examined in greater detail.
Dataset
Searching dbGaP
All publicly released dbGaP studies can be queried from the search box on the top of the dbGaP homepage. Queries can be very simple, just keywords of interest (“cancer”), or complex, making use of search fields and Boolean operators (“cholesterol[variable] AND phs000001”). More complex searches can be facilitated by using the “Advanced Search” which helps create queries via a web form.
There are many search fields available in dbGaP. Table 1 shows a selection of the most commonly used fields, explains what they search for, and gives an example of how the search would be formed.
Table 1.
This table lists fields in the dbGaP advanced search that are likely to be useful to most searchers.
Additionally, complex queries can also contain Boolean operators. For example:
Cancer[Disease] AND True[Study Has SRA Components]
returns a list of all studies having SRA data and where the PI has assigned the keyword “cancer” as a disease term.
As with all other NCBI resources, the searches in dbGaP are performed using the Entrez search and retrieval system. Please see the Entrez chapter of the NCBI handbook for general guidance on forming Entrez queries.
Once a search query is executed and results returned (Figure 3), clicking on an item’s name or accession will lead to a page listing more specific information about that object. This information is of particular importance to those users who want to find out more about a study before deciding whether or not to apply for Authorized Access. (Note that on each of the different pages, one can examine other objects in the study by using the navigational aid along the right-hand edge of the page.)
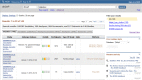
Figure 3.
This shows the returns for a simple search on the word "diabetes". Note that specific results for Studies, Variables, Study Documents, Analyses, and Datasets can be accessed by choosing the appropriate tab.
Variables on the public website
Phenotype variables can either be found by doing a search on the dbGaP home page, and then linking to an individual variable page (see Figure 4 and Figure 5 for examples), or they can be found by choosing the “Variables” tab if you are already looking at the website of a study. If you are using the “Variables” tab the phenotypes are generally grouped into broad categories for ease of browsing. These categories can be found to the right-hand side of any variable report web page (see Figure 4). Most studies use the following categories, as appropriate:
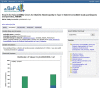
Figure 4.
Top portion of the variable report for the variable phv000200829, CARDIOV. Variables can be browsed by category using the navigation to the right hand side of the page.

Figure 5.
Top portion of the variable report for the variable phv000200829, CARDIOV. This shows how variables are linked to appropriate sections of documents. If you follow the first link, you will be taken to the portion of the document show in Figure 6.
- Affection Status
- Sociodemography and Administration
- Medical History
- Physical Observations
- Lab Measurements
- Psychological and Psychiatric Observations
- Lifestyle and Environment
- Treatment
Exceptions to this grouping method are found in large studies such as the Framingham Cohort which have their own long-standing system for grouping data. When searching for variables in large studies, or if you have a very specific query, it can be more efficient to search for variables using the search box on the right side of the variable report page (or from the dbGaP home page if you want to perform a cross-study search), rather than attempting to browse through the hierarchy of folders.
Documents on the public website
There are multiple pathways to find documents through the dbGaP web site. On the dbGaP home page the newest studies are listed under the “Latest Studies” heading, with the most direct route to documents being the orange "D" icons. A gray icon means there are no documents associated with the study. Beneath that section, the "List Top Level Studies" link leads to a searchable listing of all studies and documents, with an advanced search option available for building document-specific queries. On a study page, clicking the Documents tab will open the study's default document, with a folder tree on the right to explore the rest, and a "Search Within This Study" box that will search document text (Figure 5). Variable pages may also link to documents in which they are annotated, through the "See document part in context" links. Documents for each study are also available on the dbGaP ftp site as a downloadable zip file, which includes the pdfs, xml, and images. The ftp site is accessible from study pages by clicking the link under "Publicly Available Data."
Using document annotation
As noted previously, curators establish connections between appropriate portions of text and other accessioned objects. As an example of the types of functionality that annotations can provide, imagine looking at a variable report page having to do with whether subjects take a multivitamin. If you scroll down to the bottom of the variable page, there is a section labeled “Document parts related to the variable” which is shown in Figure 5.
This shows that there are two documents, a Coding Manual and an Annotated Form, that have text which has been associated to the multivitamin variable. If you click on the “See document part in context” link you will be taken to the appropriate portion of the document. If you follow the link for the Annotated form, you would get taken to a page that looks like Figure 6.

Figure 6.
This show the portion of the web page for the document “GoKinD Study Diabetic Offspring” (phd000152.2) which you would be taken to if you clicked the first link from Figure 5.
In the image of the questionnaire shown in Figure 6, the icon of a red circle with a white question mark,  , indicates that a section of the document is associated with one or more accessioned objects in a study. The accessioned objects are generally variables, but can include data tables. Clicking on the icon will either take the user to a variable report page (if only a single variable is associated with the icon) or to an Entrez search result page (if there are multiple objects associated with that icon).
, indicates that a section of the document is associated with one or more accessioned objects in a study. The accessioned objects are generally variables, but can include data tables. Clicking on the icon will either take the user to a variable report page (if only a single variable is associated with the icon) or to an Entrez search result page (if there are multiple objects associated with that icon).
dbGaP ftp site
The ftp site includes a directory for every study, which contains a directory for every version of a study, as well as a directory where analyses are found. Currently, each version of a study contains directories for documents, phenotype variable summaries, manifests, and release notes (Figure 7). Manifests describe the files available in each consent category while release notes describe the history of the released files as well as giving details of any changes made from previous versions.
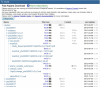
Figure 7.
This figure shows the basic organization of the ftp site, with the study phs000001.v3.p1 opened up to show portions of the hierarchy of directories and files.
Please note that older versions of studies may have a different directory structure although they contain similar information.
The variable summaries and the data dictionaries are delivered as XML files with an accompanying XSL file which produces the HTML rendering of the file that can be viewed on a browser.
The documents directory contains at least one .zip file that holds the xml files, images, and pdf versions of the documents in a study. In cases where there are a large number of documents the files may be separated into separate .zip files for xml, images, and pdf.
dbGaP Authorized Access
Data distribution by dbGaP is governed by the NIH’s policies and procedures for managing Genome Wide Association Study (GWAS) data. Information related to these policies can be found on the NIH GWAS website. Questions related to GWAS policy can be directed to vog.hin.liam@SAWG.
The individual level data is only available to authorized users. Requests for data and data downloads are managed through the dbGaP Authorized Access System (dbGaP-AA), a web platform that handles request submission, manages reviewing and approval processes carried out by signing officials (SOs) and Data Access Committees (DACs), and facilitates secured, high speed downloads of large data sets for approved users.
The dbGaP data are organized and distributed by consent groups. That is, the data are grouped by subjects that have agreed to the same set of data use limitations. The data can only be selected by consent group when making data access requests. There are no overlapping subjects between the consent groups within a study. The data requests are also reviewed and approved by consent group. Therefore it is very important that requesters understand the Data Use Limitations of consent groups before they apply for dbGaP data access.
Each data file distributed through the dbGaP has an embargo release date. The data access policy requires that the results obtained from analyzing the dbGaP data are not published before the embargo release date.
To access the Authorized Access system, non-NIH users must have an NIH eRA Commons account with a Principal Investigator (PI) role. The login username and password of a user’s dbGaP-AA account are the same as those of a user’s eRA account. NIH users need to be registered in the dbGaP system by the GWAS Project Administrator (GPA) of an affiliated institute before gaining access to the dbGaP-AA. After being registered, the NIH user can login to the dbGaP-AA account using the login username and password of their NIH CIT (or email) account.
A data access request (DAR) is made by filling out forms inside dbGaP-AA. The request includes a Research Use Statement and a Non-technical Summary. The DAR must also designate an institutional Signing Official and IT director for their project. If any of requested datasets has an IRB (Institutional Review Board) approval requirement, an IRB approval document should be uploaded to the system before submitting the request. By signing the application form, the data requester agrees to obey terms and conditions laid out in the governing Data Use Certification (DUC) document.
The DAR will first be reviewed by the SO. If approved, it will be passed on to the appropriate Data Access Committee or committees. A DAC is a committee appointed by an NIH institute (or group of institutes) which evaluates DARs requesting access to studies from their portfolio. Each DAC evaluates whether requests conform to NIH policies and procedures including whether the proposed research is consistent with the Data Use Limitations stipulated for each study. If approved, the requester must agree to obey data use restrictions dictated by participant informed consent agreements and to comply with data use, sharing, and security policies laid out in a governing DUC. At that point the data can be downloaded by the requester.
The dbGaP system manages data downloads using Aspera, a system designed to expedite high-speed data transfers. Use of Aspera requires that Aspera Connect, a browser plugin available through the Aspera website, is installed on the downloading machine. Data download can be carried out through either Aspera Connect’s web-interface or by using Aspera ASCP on the command line. For SRA (Sequence Read Archive) data distributed through the dbGaP data download can be done directly through the sra-toolkit, which allows transfer based on http protocols. Detailed information about sra-toolkit can be found from the SRA toolkit documentation.
All data downloaded from the dbGaP are encrypted. The downloaded data, with the exception of SRA data, need to be decrypted before being used. For SRA data, we suggest that users work directly with the data dump utilities that are available through the NCBI sra-toolkit without decryption. The NCBI decryption tools and sra-toolkit are available from the SRA software download site.
Approved data users are required to submit an annual project progress report to all the DACs from which they received approval. A project close-out request should be filed if the project is finished. Most dbGaP data requests have a one year approval period. To renew a project the PI needs to revise and resubmit the DAR, as well as submit the annual report. The resubmitted project will go through the SO and DAC review process again. During this process, only expired data requests under the project will be re-reviewed. Previously approved data requests that have not expired will remain approved.
A data request is not transferrable. If a PI leaves the institution listed in the DAR, all the dbGaP requests sponsored by the institution should be closed out. As a part of the close-out process, all data downloaded through the project need to be destroyed and the process has to be confirmed by the IT director and SO. The PI will need to reapply for the data once they have settled at their new location.
Related Tools
Phenotype-Genotype Integrator (PheGenI)
Scope
The Phenotype-Genotype Integrator (PheGenI), (4) merges NHGRI genome-wide association study (GWAS) catalog data with several databases including Gene, dbGaP, OMIM, GTEx and dbSNP. This phenotype-oriented resource, intended for clinicians and epidemiologists interested in following up results from GWAS, can facilitate identification and ranking of variants that may warrant additional study.
History
PheGenI was first released in 2011. The major functionality has not changed, i.e. modes of search and categories of display, but functions have been added to improve both queries and data processing. For example, an autocomplete function was added to facilitate the phenotype queries, and download functions were added to the ideogram and tabular results sections. PheGenI is under active development, with contents and displays scheduled to be more closely integrated with additional web resources.
Data Flow
PhenGenI is populated automatically via feeds from NHGRI, dbSNP, dbGaP and NCBI’s genome annotation pipeline. Please note that PheGenI does not display all p-values from each dbGaP-hosted analysis. Specifically, only p-values <10-4, and/or the lowest 100 p-values are included for each analysis. Currently, the phenotype search terms are based on MeSH, but will be enhanced with additional options in the future.
Access
Users can search based on chromosomal location, gene, SNP, or phenotype and then view and download results. Association results can be filtered by p-value, and genotype data can be filtered by location of variant site relative to gene annotation. The results are separated into several categories, including association results, genes, SNPs, eQTL data, a dynamic genome view and dbGaP studies. Each section provides a download function.
As a tool to find data in dbGaP, the view of all analysis results is accessed by clicking on the dbGaP link in the source column of the Association Results table. For full analysis and aggregate statistics such as allele frequencies, apply for controlled access.
Gene’s Phenotypes section also provides links to PheGenI, via the anchor “Review eQTL and phenotype association data in this region using PheGenI”.
The dbGaP Genome Browser
The genome wide association results hosted at the dbGaP are displayed through the dbGaP genome browser, where they can be viewed along the human genome.
The dbGaP genome browser can be accessed through the analysis page of a given dbGaP study. For example, under the “Analyses” tab of the dbGaP study phs000585.v1.p1. If there are multiple analyses, you can select one from the right panel. The link named View association results in Genome Browser leads to the chromosomal viewer and each region (block) there contains results from all tested loci within (Figure 8). The color is coded for the smallest p-value in that block.

Figure 8.
Genome Browser showing pha002914.
The genome browsing page (Figure 9) is opened by clicking on the region. The testing results are tabularized in the middle. The genome track on the top allows zooming in to see more detailed genomic location and linkage disequilibrium structure. GWAS Catalog (NHGRI) data, or an added analysis, can be aligned with the current track under the same coordinates, which allows viewers to compare results from different studies. The sequencing view (bottom) shows genome annotations (gene, transcript and protein) at that region. Each object in this page is linked to its annotated database, which helps scientists to study biological function behind the genetic variations.

References
- 1.
- Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39(10):1181–6. [PMC free article: PMC2031016] [PubMed: 17898773]
- 2.
- GAIN Collaborative Research Group. Manolio TA, Rodriguez LL, Brooks L, Abecasis G; Collaborative Association Study of Psoriasis, Ballinger D, Daly M, Donnelly P, Faraone SV; International Multi-Center ADHD Genetics Project, Frazer K, Gabriel S, Gejman P; Molecular Genetics of Schizophrenia Collaboration, Guttmacher A, Harris EL, Insel T, Kelsoe JR; Bipolar Genome Study, Lander E, McCowin N, Mailman MD, Nabel E, Ostell J, Pugh E, Sherry S, Sullivan PF; Major Depression Stage 1 Genomewide Association in Population-Based Samples Study, Thompson JF, Warram J; Genetics of Kidneys in Diabetes (GoKinD) Study, Wholley D, Milos PM, Collins FS. New models of collaboration in genome-wide association studies: the Genetic Association Information Network. Nat Genet. 2007;39(9):1045–51. [PubMed: 17728769]
- 3.
- 4.
- Ramos EM, Hoffman D, Junkins HA, Maglott D, Phan L, Sherry ST, Feolo M, Hindorff LA. Phenotype-Genotype Integrator (PheGenI): synthesizing genome-wide association study (GWAS) data with existing genomic resources. Eur J Hum Genet. 2013 [PMC free article: PMC3865418] [PubMed: 23695286]
Appendix – Phenotype Quality Control
Each submitted dataset should have a corresponding data dictionary with information describing the variables and their values. Additionally, dbGaP requires the following three special datasets to be submitted:
- 1.
Subject Consent
- 2.
Subject Sample Mapping
- 3.
Pedigree
After receiving the data submissions, QC scripts are executed to check the files for potential errors. The results are manually checked and errors are reported to submitters for clarification or resubmission of data. There are usually a few iterations prior to the study being able to be loaded.
The format of the datasets
Each dataset submitted to dbGaP should be a rectangular table showing variables in columns and subject or sample IDs in rows. Each dataset should be a single tab-delimited plain text file. Microsoft Excel files are also accepted, but are converted to tab-delimited plain text files for processing. Once the files pass all the qc checks, they are loaded into dbGaP databases and distributed as tab-delimited plain text files to approved Authorized Access users. The following formatting requirements will be ensured by running QC scripts:
- 1.
Each column has a unique, non-blank column header (variable name).
- 2.
Each dataset has a subject (or sample) ID column.
- 3.
Each row has a subject (or sample) ID value.
- 4.
There are no duplicated rows in the table.
- 5.
Datasets do not include any characters that will not be rendered correctly on the web pages.
- 6.
Duplicated subject IDs in different rows are acceptable, but will be reported in the qc checks so that curators can manually verify that there are no errors.
- 7.
Variables without any values (with only column header) are acceptable but will be reported in the qc checks.
Subject and Sample IDs
A dbGaP phenotypic dataset is a collection of variable values of individuals (subjects) or samples of individuals. Each subject should be submitted with a distinct subject ID; each sample should be submitted with a distinct sample ID. Submitters can use multiple subject alias IDs for a single subject. In addition to the submitted subject ID dbGaP assigns a single dbGaP subject ID for each submitted subject (individual person), even if the subject has multiple alias IDs. The dbGaP subject ID different from the submitted subject ID. Submitters are required to use the subject consent file to list all the subjects who participated in, or referred to by, the study, with their consent values. Subjects who did not participate in the study, namely HapMap controls and parents of participants included in the pedigree file, should also be included in the consent file with consent value 0.
The subject and sample QC scripts check the following:
- 1.
Each subject, who might be represented by multiple aliases, in the consent file has exactly one consent value (an integer).
- 2.
Each sample, which might be represented by multiple aliases, in the sample mapping file maps to exactly one subject in the subject consent file.
- 3.
All subjects in all the datasets (including subjects that have molecular data only and no phenotype data and relatives in the pedigree file) are included in the subject consent file. Additional subjects who are in the subject consent file, but are not found in any of the phenotype datasets are flagged and reported, but are not considered an error if the subject’s data will be submitted at a later time.
- 4.
Samples that are not found in the molecular data, but are found in the subject sample mapping file will be flagged and reported, but are not considered an error if the sample will be submitted at a later time.
- 5.
Multiple alternate names (aliases) for a single subject within a single or across multiple studies is assigned only one dbGaP subject ID. If the subject does not have a dbGaP subject ID, a unique ID will be assigned to the subject when the dataset is loaded into the database. Currently, a single dbGaP subject ID is assigned to a Subject only when the submitter provides the linking information. This is true at the sample level as well. The case of alternate names for samples should be less common than subjects, since dbGaP considers sample IDs to refer to the final analyte (DNA/RNA) that is put in the machine or on the chip, rather than the physical result of obtaining a tissue or blood sample, though this is accepted as well.
- 6.
If the gender of a subject is reported in multiple places (different datasets or different rows of the same dataset) the gender values should be the same.
- 7.
If a subject, or an alias of this subject, is already found in the dbGaP database, the gender of the subject in the dataset should be the same as that in the database.
- 8.
If a sample, or an alias of this sample, is already found in the dbGaP database, the sample in the dataset should map to the same subject as in the database.
- 9.
Each subject within a single study should not have conflicting case-control status, especially in the scenario when the same case control variable appears in multiple datasets.
- 10.
The number of subjects and samples are consistent between iterative submissions. When the counts are different, they are reported to the submitter for confirmation or resubmission.
HIPAA violations
The datasets submitted to dbGaP should follow the Health Insurance Portability and Accountability Act (HIPAA) rules in order to protect the privacy of personally identifiable health information. Due to the complexity of HIPAA rules, it is impossible to write a program to report all HIPAA violations without turning up false positives. It is also impractical to manually check all the data values and find all the HIPAA violations. QC scripts have been created to check variable names, descriptions, and values, and to flag variables that are likely to have sensitive information. dbGaP curators then manually check the flagged variables to determine whether these are HIPAA violations. The QC scripts first report all variables whose names or descriptions contains the following key words (case insensitive except for ‘IP’ and ‘DOB’):
- 1.
name
- 2.
address
- 3.
zip
- 4.
phone
- 5.
telephone
- 6.
fax
- 7.
mail
- 8.
email
- 9.
social
- 10.
ssn
- 11.
ss#
- 12.
birth
- 13.
DOB
- 14.
license
- 15.
account
- 16.
certificate
- 17.
vehicle
- 18.
url
- 19.
IP
Only the names or descriptions containing a whole-word match with at least one of the above key words are reported. A word in the variable name or description that contains a key word as a substring is not considered a match. For example, “Leave your email/phone.” is reported as a match since it contains key words “email” and “phone”, but “zipper” is not reported because it only contains key word “zip” as a substring.
In many cases the variable names or descriptions do not have any indication that the variable might have HIPAA incompatible information. To work around this, the QC scripts also check variable data values for sensitive information. Data values are much harder to check than variable names and descriptions due to the sheer number of individual values and the great variety of errors. Fortunately, almost all of the HIPAA violations in the datasets submitted to dbGaP database are related to dates, including dates as separated values and dates embedded in longer texts. Below are some of the examples of the dates found in the datasets submitted to dbGaP:
- 11-JUN-1970
- 01-SEP-65
- SEPT-NOV 1995
- 2004.05.10
- 2/2/85
- 8-11-83
- 10/1974
- JAN ‘93
- 3/04
- SEPT 85
- NOV-89-
- FEB64
- NOVEMBER 1992
- “DEC” “92”
- Feb.4
- Jan – 1996
- March, 2004
- (Nov,2005)
- APR. “85
- APRIL 91
- apr 2000
- (APRIL 1997)
- (3/00)
- DEC.1992
- 1998-May
- October-September, 2004
- Jan. 1
- May 3rd
- xxxxxxIN2002.03.01
- 19941122
- 112004
Most of the above values, e.g., “01-SEP-65”, “2004.05.10”, “DEC.11992”, are obviously date values and not HIPAA compatible. It is hard to write programs to find dates in all the different formats without generating too many false positives. However, some of them are not so obvious and need manual confirmation using variable descriptions and context of the values. For example, “3/04” could mean “March 2004”, or “3 out of 4”; 19941122 could be “Nov. 22, 1994” or the number 19941122; “112004” could be “Nov. 2004”, “Nov. 20, 2004” or the number 112004. If we report all the 6-digit numbers as potential date values, we will generate a great amount of false positives. More complicated algorithms are needed to detect date values with high sensitivity without sacrificing too much specificity. We use the following algorithm to detect the date values in the datasets:
- 1.
Two 1 or 2-digit numbers and a 2 or 4-digit number, in this order, separated by “/”, “-“ or “.”, e.g., “3/5/1994” or “12-28-03”.
- 2.
One 4-digit number and two 1 or 2-digit numbers separated by “/”, “-“ or “.”, e.g., “1994.2.13”.
- 3.
A 1 or 2-digit number and a 4-digit number starting with 19 or 20 separated by “/”, e.g., “10/1994” (but not “10.1994”).
- 4.
A 1 or 2-digit number followed by a “/” and a 2-digit number starting with 0, e.g., “3/04” (but not “3/94”).
- 5.
A month name or short name and a 1, 2, or 4-digit number, in either order, separated by some non-letter, non-number characters or not separated, e.g., “JAN ‘93”, “FEB64”, “May 3rd” (but not “may be 14”). An example of a false positive is “4 (may be under reporting)”.
- 6.
A 6-digit number is considered to be a potential date value if its first four digits make a valid date in mmdd format (i.e., first two digits read as month second two as day of the month). For example, 122876 is considered to be a potential date value since 1128 is a valid date (Nov. 28) in mmdd format; 231208 or 113198 is not a potential date since 2312 or 1131 is not a valid date in month/day format. If all of the values, or first 10 values, of a variable are 6-digit potential dates, this variable together with its potential date values will be reported by the scripts.
- 7.
An 8-digit number is considered to be a potential date value if it makes a valid date in the 20th or 21st century in either mmddyyyy or yyyymmdd format. For example, 19940822 is considered to be a potential date since it can be read as 1994/08/22 (Aug. 22, 1994). 10312005 is a potential date value since it can be read as 10/31/2005 (Oct. 31, 2005). “19080230” is not considered to be a potential date since neither 1908/02/30 nor 19/08/0230 is a valid date in the 20th or 21st century. If all of the values or the first 10 values of a variable are 8-digit numbers of potential date values, the variable will be reported as containing potential HIPAA violations.
In addition, the QC scripts also report values that look like social security numbers (e.g., “123-45-6789” or “123456789”), phone numbers (e.g., “321-456-7890” or “(301)456-7890”), zip codes (e.g., “MD 20892”), etc. A few cases of this kind of information have been detected by the QC scripts. However, other cases like names of people are not found by the QC scripts, but by human curation.
Extreme values that might be used to identify individual participants (ages over 90, extremely heavy body weights, families with extraordinary large numbers of children) are also HIPAA violations. The QC scripts infer age variables from variable names, descriptions, and units and report ages over 89 as potential HIPAA violations. For other extreme values, since the HIPAA rules don’t specify particular cut-off values, we check the value distribution curves by hand and decide whether we need to hide the extreme values on a case-by-case basis.
Data dictionaries
Data dictionaries are required to be submitted along with every dataset, to explain the meanings of the variables and data values. For each value in the datasets, dbGaP requires that the submitters provide a variable description, variable type, units of values for numerical variables, as well as logical minimum and maximum values if available. For each encoded value, a code meaning should be included in the data dictionary. Since the data dictionaries submitted to dbGaP vary in format, many of which are not quite machine-readable, curators spend a good deal of time understanding the data dictionaries, correcting errors, and making other modifications so that they can be read by computer programs. Then QC scripts are executed to compare the data dictionaries with the corresponding datasets. The QC scripts report variables in the datasets that are missing required information (such as descriptions) in the data dictionary, as well as variables described in the data dictionaries but not found in the datasets. Many of these mismatches are caused by typos, such as “0” for “O” and vice-versa. A number of the numerical variables submitted to dbGaP are missing units. Often the units are implied in a variable’s description or in other documents. The QC scripts try to add the units back by checking the variable descriptions, which is then verified by manual curation.
In addition to missing descriptions and units, many datasets submitted to dbGaP have missing, or incomplete, code/value pairs. Some of these errors are easy to detect, e.g., the values of a categorical variable are encoded by integers and all of the code meanings are provided except for one code. However, if the variables contain both numerical values and numerically encoded categorical values, the errors of missing code meanings are hard to detect. Usually in this case, the submitters would use numbers beyond logical value range to encode for non-numerical meanings. For example, if the variable is age of patient, they would use code values like -1, 999 for meanings like “N/A” or “unknown”. If the submitters provided logical minimum and maximum values to us, it would be easy for us to find all the missing code meanings. However, in most of the cases the logical minimum and maximum values are either not available or incorrect. Unreasonable or suspicious values found automatically or manually are reported to submitters to clarify and correct.
QC scripts are executed to compare each submitted dataset to its corresponding data dictionary. The QC scripts report the following errors or potential errors:
- 1.
Variables missing descriptions in data dictionary.
- 2.
Variables with descriptions in data dictionary but not found in dataset (usually due to manual typos in variable names).
- 3.
Potential errors or missing information in value code meanings.
The following algorithm is used to detect potential errors in the code meanings of each variable:
- 1.
If the variable is labeled as code-value type by the submitter, report all values in dataset without code meanings in data dictionary.
- 2.
If all the values are numbers,
- i.
Report extreme values beyond 5×SD as potential encoded values. Exclude 5 largest numbers when calculating SD.
- ii.
Report rare negative numbers as potential encoded values. A negative number is considered to be rare if only 1 or 2 out of total more than 10 distinct values, or less than 1% of the distinct values are negative numbers.
- 3.
If all the values are non-number texts,
- i.
Report all the values without code meanings in the data dictionary if more than half of the distinct values have code meanings.
- ii.
Report all the values in the dataset that differ from a code in the data dictionary only by case. For example, if the data dictionary includes code meaning “UNK=Unknown”, but the dataset has a value “Unk” instead of “UNK”, the scripts report the case mismatch.
- 4.
If some of the values are numbers but some are non-numbers, separate the values into a set of numerical values and a set of text values, then report the potential encoded values using the above rules.
- 5.
Report all the code values in data dictionary but not used in the dataset.
Again there is a trade-off between sensitivity and specificity. The QC scripts allow the curator to set some parameters like cut-off number of SDs to adjust the sensitivity and specificity. For example, if too many real extreme values are reported as potential encoded values, i.e., the false positive rate is high, we can set the parameter to let the QC scripts report only the extreme values beyond 6×SD or more.
Pedigree file
If there are related individuals in the study, a pedigree file should be submitted to dbGaP. Pedigrees can be quite complex depending on the number of vertical and horizontal relationships, however all relationships can be summarized using the following five required columns: family ID, subject ID, father ID, mother ID, and sex. dbGaP also collects twin IDs, where these IDs can be expanded to include multiples. An additional column can be included to differentiate monozygotic and dizygotic twins, and twin ID if available. All subjects who appear in the father ID or the mother ID columns should also be included in the subject ID column. QC scripts were created to check the pedigree files and report the following errors or potential errors:
- 1.
Any of the above required columns is missing.
- 2.
Subject IDs appearing more than once in the subject ID column.
- 3.
Father or mother IDs that are not found in the subject ID column.
- 4.
Subjects missing family IDs.
- 5.
Subjects missing sex values.
- 6.
Male subjects shown in the mother ID column and female subjects as fathers.
- 7.
Subjects with non-null but same father and mother IDs.
- 8.
Subjects having children with their parents or grandparents.
- 9.
Subjects having children with their sibling or half siblings.
- 10.
Subjects having children with their uncle or aunts.
- 11.
Subjects having children with their cousins (Usually these are not errors. We flag them out just to make sure the data is correct.)
- The Database of Genotypes and Phenotypes (dbGaP) and PheGenI - The NCBI HandbookThe Database of Genotypes and Phenotypes (dbGaP) and PheGenI - The NCBI Handbook
Your browsing activity is empty.
Activity recording is turned off.
See more...