Clinical Description
MBTPS1-related spondyloepimetaphyseal dysplasia with elevated lysosomal enzymes (MBTPS1-SEMD) is characterized by postnatal-onset short stature, pectus deformity, kyphosis and/or scoliosis, hernia, protruding abdomen, cataract(s), developmental delay, and dysmorphic facial features, in combination with elevated lysosomal hydrolase enzyme levels in plasma. Additional features can include waddling or staggering gait, craniosynostosis, and seizures. To date six individuals with MBTPS1-SEMD from six families have been reported [Kondo et al 2018, Carvalho et al 2020, Meyer et al 2020, Alotaibi et al 2022, Chen et al 2023, Yuan et al 2023]. Three additional affected individuals are known to the authors. The features in Table 2 and the following description are based on published reports.
Growth deficiency. Postnatal-onset short stature was present in all reported individuals and was typically identified by age three years. Two individuals were treated with growth hormone therapy. One individual started treatment at age three years and discontinued after one year without improvement in growth velocity [Kondo et al 2018]. In a second individual with normal insulin-like growth factor 1 (IGF-1), recombinant intravenous growth hormone (rhGH) of 0.15-0.2 IU/kg per day was started at age three years; after three years of rhGH therapy, the height increased from 5.3 standard deviations (SD) below the mean (before treatment) to 3.96 SD below the mean (after three years of treatment) [Chen et al 2022]. The range of severity of growth deficiency following treatment with growth hormone was from 2.9 to 5.3 SD below the mean.
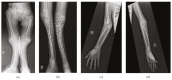
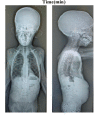
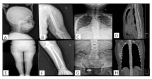
Musculoskeletal manifestations. Chest deformity including pectus carinatum, pectus excavatum, or unspecified sternal malformation was reported in all individuals. Kyphoscoliosis or scoliosis was present in all individuals. Reduced bone density was evident in all individuals. Anterolisthesis of L5 on S1 has been described in one individual [Kondo et al 2018].
Hip dislocation with coxa vara was reported in one individual [Alotaibi et al 2022]. Waddling or staggering gait was reported in two individuals [Alotaibi et al 2022, Yuan et al 2023].
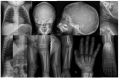
Craniosynostosis was identified in two individuals [Carvalho et al 2020], including one individual originally reported by Kondo et al [2018] with craniosynostosis identified after images were reevaluated by the authors.
Short neck and rhizomelia were reported in one individual [Carvalho et al 2020]; brachydactyly, genu valgum, valgus tibial bowing, and pes cavus were reported in one individual [Alotaibi et al 2022]; pes valgus and prominent sandal grooves were reported in one individual [Meyer et al 2020] (see ). Hyperextended fingers were reported in one individual [Yuan et al 2023].
Hernia or protruding abdomen were reported in all individuals. Bilateral inguinal hernia was reported in two individuals [Kondo et al 2018] and unilateral inguinal hernia with protruding navel in one individual [Meyer et al 2020]. Three individuals had protruding abdomen [Carvalho et al 2020, Alotaibi et al 2022, Chen et al 2022].
Cataracts were common. Two individuals had congenital lamellar cataract, with surgical lens removed at age ten months [Carvalho et al 2020] and age 35 months [Chen et al 2022]. One individual had punctiform opacities of the lens identified at age 11 years, keratoconus that was treated at age 11 years, and anterior and posterior subcapsular cataract that was detected at age 18 years [Meyer et al 2020].
Developmental delays. Most individuals had gross motor delays. Three individuals started walking at age 15 months, 24 months, and 36 months [Carvalho et al 2020, Meyer et al 2020, Alotaibi et al 2022]. Three individuals had speech and language delay [Meyer et al 2020, Alotaibi et al 2022]; one of these children had first sounds at age 21 months and feeding problems with poor weight gain managed with gastrostomy tube feeding for several months [Meyer et al 2020]. One individual was reported to have expressive language delay, with first words spoken at age 26 months [Carvalho et al 2020].
Intelligence is normal in the majority of individuals. One individual was reported with below-average intelligence [Alotaibi et al 2022], and a second individual had mild intellectual disability with an IQ of 57 [Yuan et al 2023].
Dysmorphic features. Characteristic dysmorphic features were present in the majority of individuals, including prominent forehead, prominent cheekbones, retromicrognathia, wide mouth, and large, prominent ears (see ).
Other features
Accumulation of fat on the chest and abdomen (1 individual) [
Yuan et al 2023]