Clinical Description
Mucopolysaccharidosis type IVA (MPS IVA) comprises a clinical continuum ranging from a severe and rapidly progressive form to a slowly progressive form. In the past, the two forms were distinguished by height, the subjective assessment of severity of bone deformity, and survival [Tomatsu et al 2011]; however, neither clinical nor biochemical findings can provide clear distinctions between the two forms and, thus, the MPS IVA phenotype should be considered a continuum from severe to slowly progressive.
Some features including prominent forehead, pectus carinatum, kyphosis, and abnormal spine radiograph may be detected at birth [Peracha et al 2018]. However, the majority of infants have no distinctive clinical findings. The severe form is usually apparent between ages one and three years. The slowly progressive form may not become evident until late childhood or adolescence [Montaño et al 2007].
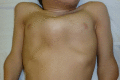
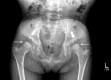
In both the severe form and slowly progressive form, the initial presentations vary and individuals may present with only a single finding or several findings. Kyphoscoliosis, genu valgum (), and pectus carinatum ( and ) are the most common initial manifestations of the severe form [Montaño et al 2007]. In contrast, hip problems including pain and stiffness (due to collapse and flattening of the proximal femoral epiphysis) are common initial manifestations of the slowly progressive form [Hecht et al 1984, Wraith 1995, White et al 2014].
Because descriptions of the natural history of MPS IV published in the past may not have distinguished between MPS IVA (Morquio syndrome type A; accounting for >95% of affected individuals) and MPS IVB (Morquio syndrome type B; <5% of affected individuals), the following information is relevant to both MPS IVA and MPS IVB.
While the skeletal findings of MPS IVA are the hallmark findings, involvement of other organ systems can lead to significant morbidity, including respiratory compromise, obstructive sleep apnea, valvular heart disease, hearing impairment, corneal clouding, dental abnormalities, and hepatomegaly. Compression of the spinal cord results in neurologic involvement especially when disease is recognized later in life [Neufeld & Muenzer 2001, Tomatsu et al 2011, Solanki et al 2013].
Coarse facial features are also present but milder than in other mucopolysaccharidoses (see Differential Diagnosis).
Children with MPS IVA typically have normal intellectual ability.
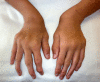
Ligamentous laxity and joint hypermobility are distinctive features of MPS IVA, and are rare among other storage disorders.
Musculoskeletal
Skeletal findings worsen over time. The combination of bone and joint involvement leads to pain and arthritis that result in subsequent disability.
Upper-extremity involvement is also progressive and can impair hand-wrist strength and limit ability to perform some activities of daily living, such as using a fork. Hypermobility and ulnar deviation of the wrist joint due to premature cessation of ulna growth are distinctive features of MPS IVA.
Lower-extremity involvement, which is universal and progressive if untreated [Holzgreve et al 1981, Dhawale et al 2012], generally consists of malalignment due to progressive hip subluxation and/or valgus deformity. It can lead to significant gait alteration; hip, knee, and/or ankle pain with activity; and decreased endurance [Dhawale et al 2012, Peracha et al 2018].
Knee and ankle valgus are the most common lower-extremity deformities. Genu valgum (knock-knee) results from distal femoral and proximal tibial involvement and laxity of the collateral ligament.
A longitudinal study using the Pediatric Evaluation of Disability Inventory and the Functional Independence Measure found severely limited joint mobility in persons with MPS IVA, generally with loss of ambulation late in the disease course. Aggressive and long-term intervention by a team of physical therapists and rehabilitative specialists is often needed to optimize mobility (see Management) [Guarany et al 2012].
Hip dysplasia. Early in the disease course, the capital femoral epiphyses are small and the acetabula are shallow. Subsequent progressive flattening and fragmentation of the capital femoral epiphyses in the femoral head and acetabular dysplasia result in hip dislocation, arthritis, and severe joint restriction, causing affected individuals to become wheelchair bound [Tomatsu et al 2011, Peracha et al 2018].
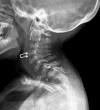
Spinal cord compression may occur in any spinal segment; cervical spinal compression is the most common site. Spinal cord compression can be caused by cervical instability, unossified fibrocartilage associated with an abnormal odontoid process, ligamentous laxity, cartilaginous and ligamentous hypertrophy at the atlantoaxial joint, glycosaminoglycan (GAG) deposition in the extradural space, disc protrusion, thoracolumbar kyphosis, and acquired central canal stenosis [Lipson 1977, Ransford et al 1996, Tomatsu et al 2011, McKay et al 2012, Solanki et al 2013].
Odontoid hypoplasia leading to atlantoaxial instability, which later may result in upper cervical spinal cord compression, occurs in 90% of affected individuals () [Hughes et al 1997]. Persons with untreated atlantoaxial instability often do not survive beyond the second or third decade because minor falls and/or neck extension can result in quadriparesis or sudden death [Tomatsu et al 2011].
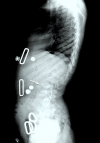
Spinal canal stenosis may be diffuse or focal. The causes of spinal canal stenosis are similar to the causes of spinal cord compression and include: kyphosis, disc protrusion, generalized thickening of posterior longitudinal ligament, or localized thickening of intervertebral ligaments due to GAG deposition.
Lumbar spine malalignment (i.e., thoracolumbar kyphosis) in older individuals can result in focal spinal stenosis, compressive myelopathy, and paraplegia [Dalvie et al 2001].
Ligamentous laxity resulting in hypermobile joints is common; however, decreased joint mobility can be observed in the large joints including knees, hips, and elbows [Neufeld & Muenzer 2001].
Neurologic
At the time of diagnosis, individuals typically have normal developmental milestones and normal intellectual ability. The neurologic findings of MPS IVA are most often secondary to spinal abnormalities in the neck and/or lumbar region. The increased risk for neurologic compromise makes developmental delay and learning disabilities more common in children with MPS IVA than in unaffected children [Montaño et al 2007].
Subtle abnormal brain MRI findings such as prominent perivascular space, enlarged lateral ventricles, and prominent frontal CSF were reported in eight of 14 individuals with MPS IVA [Davison et al 2013]. From the same study, neurocognitive evaluation revealed behavioral issues including anxiety, depression, decreased attention span, and somatic complaints. Whether these findings are caused by disease-specific biochemical abnormalities, chronic illness, or a combination of the two is unknown.
Cardiac
Cardiac complications include ventricular hypertrophy and early-onset severe valvular involvement. Coronary intimal sclerosis has also been reported [Hendriksz et al 2013].
In a multicenter, multinational, cross-sectional study (MorCAP) involving 325 individuals with MPS IVA, valvular regurgitation was more common than valvular stenosis. Among those with valvular regurgitation, tricuspid regurgitation was the most common (35%). Mitral regurgitation, aortic regurgitation, and pulmonary regurgitation were found in 25%, 19%, and 14% of affected individuals, respectively [Harmatz et al 2013]. Individuals with MPS IVA usually have an abnormally age-related aortic root dilatation, high heart rate and high myocardial index to compensate for small left ventricular diameter, impaired diastolic filling, and low stroke volume [Hendriksz et al 2015, Akyol et al 2019].
Respiratory
Respiratory complications are a major cause of morbidity and mortality. Airway obstruction, sleep-disordered breathing, and restrictive lung disease have been described.
GAG accumulation in the adenoids, tonsils, pharynx, larynx, trachea, and bronchial tree leads to adenotonsillar hypertrophy, tracheal distortion, tracheo- and bronchomalacia, and obstructive sleep apnea [Semenza & Pyeritz 1988, Walker et al 2003]. Deposition of GAGs in the trachea and bronchi could cause tortuosity of the airway leading to buckling and airway obstruction when the neck is flexed. If not recognized (particularly during cervical spine fusion/stabilization surgery) and if the head and neck are fused in a flexed position, acute obstruction may result in tracheal extubation [Solanki et al 2013]. In addition to tracheal narrowing from GAG accumulation, external factors including brachiocephalic artery crossing the anterior of the trachea and small thoracic inlet also contribute to tracheal stenosis [Peracha et al 2018].
Because of atlantoaxial instability and upper-airway obstruction, persons with MPS IVA prefer to sleep prone on a flat surface without a pillow in order to keep the neck extended and minimize the tortuosity of the airway.
Restrictive lung disease results from a small thorax, chest wall anomalies, spine deformities, neuromuscular compromise from cervical myelopathy, and hepatomegaly causing upward displacement of the diaphragm [Walker et al 2003, Hendriksz et al 2013].
If respiratory complications are not recognized or are not treated, cor pulmonale and respiratory failure can ensue, leading to early death [Hendriksz et al 2013, Peracha et al 2018].
Growth
Growth and final height are used as an indicator of severe phenotype. Children with MPS IVA have a normal birth weight and a longer-than-normal birth length. The growth velocity decreases between ages one and three years and growth stops around ages seven to eight years [Montaño et al 2007, Peracha et al 2018]. By age 18 years, the average height in males is 123 cm and in females 117 cm, compared to 177 cm and 163 cm in unaffected males and females, respectively.
Eye
Ophthalmologic findings are present in more than 50% of individuals with MPS IVA. Natural history studies have not been performed; thus, it is not possible to predict the age of onset of ophthalmologic findings [Wood et al 2013].
Slowly progressive corneal clouding, found in 50% of affected individuals (ages 1-65 years), is the most common ophthalmologic finding in MPS IVA.
Other less common ophthalmologic findings include: astigmatism, cataracts, punctate lens opacities, open-angle glaucoma, optic disc swelling, optic atrophy, and retinopathy. While rare, these ophthalmologic findings can be serious secondary complications [Hendriksz et al 2013, Hendriksz et al 2015].
Pseudoexophthalmos, the appearance of a bulging eye secondary to a shallow orbit, can cause exposure keratitis and also be of cosmetic concern.
Dental
Deciduous teeth erupt normally and are widely spaced and discolored with thin irregular (stippled) enamel and small pointed cusps which flatten over time with normal wear.
Permanent teeth also have hypoplastic enamel, and are widely spaced with flared upper incisors [Onçağ et al 2006, Peracha et al 2018].
Hearing
Mild-to-moderate hearing loss is common in individuals with MPS IVA. Hearing impairment is often noted toward the end of the first decade.
Mixed (i.e., combined conductive and sensorineural) hearing loss is more common than conductive or sensorineural hearing loss alone.
Conductive hearing loss is secondary to recurrent middle ear infections, serous otitis media, and deformity of the ossicles [Hendriksz et al 2013].
Sensorineural hearing loss secondary to GAG accumulation in the inner ear and/or central nervous system has been described [Ruckenstein et al 1991, Simmons et al 2005, Hendriksz et al 2013].