Summary
Clinical characteristics.
Multicentric osteolysis nodulosis and arthropathy (MONA) is a skeletal dysplasia characterized by progressive osteolysis (particularly of the carpal and tarsal bones), osteoporosis, subcutaneous nodules on the palms and soles, and progressive arthropathy (joint contractures, pain, swelling, and stiffness). Other manifestations include coarse facies, pigmented skin lesions, cardiac defects, and corneal opacities. Onset is usually between ages six months and six years (range: birth to 11 years).
Diagnosis/testing.
The diagnosis of MONA is established in a proband with characteristic clinical and radiographic findings and either biallelic pathogenic variants in MMP2 or decreased activity of the enzyme matrix metalloproteinase 2.
Management.
Treatment of manifestations: Treatment is supportive only and can include physical therapy (which may slow the rate of development of contractures and prolong mobility) and mobility aids. Pain medications may not provide relief unless the pain is due to secondary osteoarthritis/contractures. Daily recommended supplementation of vitamin D and calcium; standard management of congenital heart defects; management of scoliosis and kyphosis per orthopedist.
Surveillance: Annual joint assessment by a rheumatologist or orthopedic surgeon. Evaluation with cardiologist as needed for follow up of congenital heart disease. Annual assessment for scoliosis and kyphosis.
Agents/circumstances to avoid: Physical trauma to reduce the risk of fractures.
Genetic counseling.
MONA is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the MMP2 pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives and prenatal and preimplantation genetic testing for a pregnancy at increased risk are possible.
Diagnosis
Formal diagnostic criteria have not been established for multicentric osteolysis nodulosis and arthropathy (MONA).
Suggestive Findings
MONA should be suspected in individuals with the following clinical findings, radiographic findings, and family history.
Clinical findings
- Subcutaneous nodules, usually on the palms and soles (See Figure 2.)
- Coarse facial features
- Gingival hypertrophy


Radiographic findings
- Progressive osteopenia and osteolysis with early and predominant involvement of the bones of the hands (see Figure 4) and feet (see Figure 5). The carpal and tarsal bones, which are initially well formed, slowly show thinning of the cortices, osteopenia, and finally osteolysis, becoming small and irregular or disappearing completely.
- Thin cortices of the long bones (see Figure 6). The metacarpals, metatarsals, and phalanges are most affected.
- Milder and similar changes in other bones
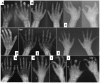
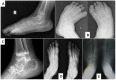
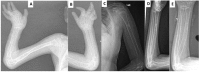
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis
Establishing the Diagnosis
The diagnosis of MONA is established in a proband with characteristic clinical and radiographic findings by identification of biallelic pathogenic (or likely pathogenic) variants in MMP2 on molecular genetic testing (see Table 1) or decreased activity of the enzyme matrix metalloproteinase 2 by gelatin zymography.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic MMP2 variants of uncertain significance (or of one known MMP2 pathogenic variant and one MMP2 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular Genetic Testing
Testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of MONA has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
- Single-gene testing. Sequence analysis of MMP2 is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
- A multigene panel that includes MMP2 and other genes of interest (see Differential Diagnosis) may also be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2. Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Multicentric Osteolysis Nodulosis and Arthropathy
Analysis of Enzyme Activity
Analysis of matrix metalloproteinase 2 enzyme activity can be performed by the electrophoretic technique gelatin zymography on body fluids and/or cell or tissue extracts [Martignetti et al 2001, Azzollini et al 2014]. In contrast to controls, affected individuals show complete loss of enzyme activity.
Clinical Characteristics
Clinical Description
Multicentric osteolysis nodulosis and arthropathy (MONA) is a skeletal dysplasia characterized by progressive osteolysis (particularly of the carpal and tarsal bones), osteoporosis, subcutaneous nodules on the palms and soles, and progressive arthropathy (contractures, pain, joint swelling/stiffness). Other manifestations include pigmented lesions on the skin, coarse facies, corneal opacities, and cardiac defects.
Most affected children are apparently normal at birth. Onset is usually between ages six months and six years [Azzollini et al 2014]; the range is from birth to 11 years [Castberg et al 2013, Bhavani et al 2016].
To date, 51 individuals have been identified with biallelic pathogenic variants in MMP2 [Martignetti et al 2001, Zankl et al 2005, Rouzier et al 2006, Phadke et al 2007, Zankl et al 2007, Tuysuz et al 2009, Gok et al 2010, Jeong et al 2010, Temtamy et al 2012, Castberg et al 2013, Azzollini et al 2014, Ekbote et al 2014, Bader-Meunier et al 2016, Bhavani et al 2016, Pichler et al 2016, Kröger et al 2019, Elsebaie et al 2021]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Multicentric Osteolysis Nodulosis and Arthropathy: Frequency of Select Features
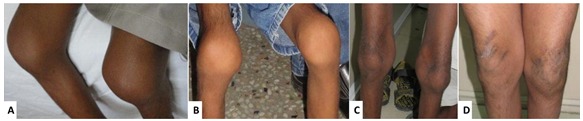
Joint manifestations. Peripheral joints are more involved than proximal joints. Small joints of the hands and feet are the most obvious sites of involvement. Progressive bone destruction leads to pain, swelling, stiffness, and later flexion contractures (Figure 1, Figure 2). Foot and hand deformities and progressive shortening of digits occur with age.
Knees show swelling and contractures in the majority (Figure 3); hip involvement is less severe [Bhavani et al 2016].

Figure 3.
Swollen knee joints in children with MONA at age 6 years (A), 7 years (B), and 13 years (C,D). Note serpiginous hyperpigmented skin lesions in C and D. From Bhavani et al [2016]; republished with permission of John Wiley and Sons
Wrists, ankles, and elbows are also involved.
Over time all affected individuals need assistance with mobility. Those with more severe manifestations are wheelchair bound between ages three and 12 years [Zankl et al 2005, Rouzier et al 2006, Temtamy et al 2012].
Osteopenia. Low bone density and osteoporosis may lead to an increased risk of fracture of long bones and vertebrae [Jeong et al 2010, Temtamy et al 2012].
Short stature. Most individuals have short stature [Castberg et al 2013]. Stature is normal in early childhood; progressive bone and joint deformities could be the cause of short stature.
Facial features. Coarsening of facial features, bulbous nose, proptosis, strabismus, and gingival hypertrophy have been observed in several affected individuals [Bhavani et al 2016].
Skin. Subcutaneous, firm, palpable nodules in the palms and soles are noted in the majority of (but not all) affected individuals (Figures 2B and 2C). The presence of subcutaneous nodules may be age dependent. To date it has not been possible to determine if these nodules are painful as affected individuals have pain in their hands and feet due to osteopenia, fractures, contractures, limitation of movement, and sometimes inflammation.
Other cutaneous manifestations include hyperpigmentation and thickening. Serpiginous hyperpigmented cutaneous lesions are present in a few individuals (Figures 3C and 3D) [Zankl et al 2005, Azzollini et al 2014, Bhavani et al 2016].
In some individuals excessive skin folds in the hands and feet have resulted from destruction of underlying bones (Figure 1I).
Heart. Congenital heart defects (transposition of great arteries, atrial septal defect, ventricular septal defect, bicuspid aortic valve, and mitral valve prolapse) have been observed in one third of individuals reported to date [Tuysuz et al 2009, Castberg et al 2013, Bhavani et al 2016].
Intellect is normal.
Other
- Scoliosis and kyphosis are observed in some individuals [Temtamy et al 2012, Azzollini et al 2014, Bhavani et al 2016, Kröger et al 2019].
- Corneal opacities are occasionally observed; however, vision is preserved.
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been observed.
Nomenclature
In addition to MONA, this phenotype has been reported in the literature as Torg syndrome, Winchester-Torg (or Torg-Winchester) syndrome, and nodulosis-arthropathy-osteolysis (NAO) syndrome. All of these conditions have been shown to be caused by biallelic pathogenic variants in MMP2 with no discernible genotype-phenotype correlation. The term MONA is therefore used throughout this GeneReview to refer to all of these conditions.
In the 2023 revision of the Nosology of Genetic Skeletal Disorders [Unger et al 2023], MONA is referred to as MMP2-related multicentric osteolysis, nodulosis, and arthropathy and is included in the osteolysis group.
Prevalence
The prevalence for this rare skeletal dysplasia is not available at present. To date 51 individuals (from 31 families) with molecularly proven MONA have been reported.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in MMP2.
Sporadic tumors (including gastric, colorectal, ovarian, melanoma, breast, and lung cancers) occurring as single tumors in the absence of any other findings of multicentric osteolysis nodulosis and arthropathy frequently harbor a somatic variant in MMP2 that is not present in the germline. In these circumstances predisposition to these tumors is not heritable. For more information, see Cancer and Benign Tumors.
Differential Diagnosis
Table 3.
Disorders to Consider in the Differential Diagnosis of Multicentric Osteolysis Nodulosis and Arthropathy (MONA)
Juvenile idiopathic arthritis. Like MONA, juvenile idiopathic arthritis is characterized by joint pain, swelling, and stiffness. Features distinguishing this condition from MONA include joint inflammation (tenderness and warmth), increased erythrocyte sedimentation rate, and C-reactive protein (minimal in some instances).
Bacterial infections and tubercular osteomyelitis may show some resemblance clinically and radiologically.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with multicentric osteolysis nodulosis and arthropathy (MONA), the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Multicentric Osteolysis Nodulosis and Arthropathy
Treatment of Manifestations
Treatment is supportive.
Table 5.
Treatment of Manifestations in Individuals with Multicentric Osteolysis Nodulosis and Arthropathy
Surveillance
Table 6.
Recommended Surveillance for Individuals with Multicentric Osteolysis Nodulosis and Arthropathy
Agents/Circumstances to Avoid
Avoid physical trauma to reduce the risk of fractures.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
No information on pregnancy management and outcomes is available.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Multicentric osteolysis nodulosis and arthropathy (MONA) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., presumed to be carriers of one MMP2 pathogenic variant based on family history).
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for an MMP2 pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- One of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017].
- Uniparental isodisomy for the parental chromosome with the pathogenic variant resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for an MMP2 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- The age of onset and manifestations may vary in sibs with the same biallelic MMP2 pathogenic variants [Kröger et al 2019].
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. The offspring of an individual with MONA are obligate heterozygotes (carriers) for an MMP2 pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an MMP2 pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the MMP2 pathogenic variants in the family.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the MMP2 pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Multicentric Osteolysis Nodulosis and Arthropathy: Genes and Databases
Table B.
OMIM Entries for Multicentric Osteolysis Nodulosis and Arthropathy (View All in OMIM)
Molecular Pathogenesis
MMP2 encodes metalloproteinase-2 enzyme (MMP2), a type IV collagenase protein also known as gelatinase. MMP2 consists of four functional domains: signal peptide, propeptide domain, catalytic domain, and hemopexin domain.
Pathogenic variants include deletions and missense, nonsense, and splice site variants. Most pathogenic variants are private and homozygous. Most MMP2 pathogenic variants lead to loss of gelatinolytic activity of the enzyme MMP2 [Azzollini et al 2014].
The study of osteoblast cell lines from Mmp2-null mice showed that the loss of Mmp2 protein leads to decreased bone mineralization, destruction of articular cartilage, joint erosion, and defects in osteoblast and osteoclast growth [Mosig et al 2007].
Mechanism of disease causation. Loss of function
Cancer and Benign Tumors
Somatic MMP2 pathogenic variants that result in abnormal MMP2 expression are associated with cancers (e.g., gastric, colorectal, ovarian, melanoma, breast, and lung cancers). The COSMIC (Catalogue of Somatic Mutations in Cancer) database shows 192 unique MMP2 variants in tissues associated with cancers.
Chapter Notes
Author Notes
Dr Katta M Girisha, MD, DM is a clinical geneticist with special interest in skeletal dysplasia and genomic diagnosis of rare diseases.
Website: kmcmedicalgenetics.in
Acknowledgments
Indian Council of Medical Research – Clinical and molecular evaluation of inherited arthropathies and multiple vertebral segmentation defects (BMS 54/2/2013)
Department of Science and Technology – Application of autozygosity mapping and exome sequencing to identify genetic basis of disorders of skeletal development (SB/SO/HS/005/2014)
Revision History
- 30 March 2023 (sw) Revision: "MMP2-Related Multicentric Osteolysis, Nodulosis, and Arthropathy" added as a synonym; Nosology of Genetic Skeletal Disorders: 2023 Revision [Unger et al 2023] added to Nomenclature
- 9 September 2021 (sw) Comprehensive update posted live
- 14 July 2016 (bp) Review posted live
- 22 February 2016 (kmg) Original submission
References
Literature Cited
- Al-Mayouf SM, Majeed M, Hugosson C, Bahabri S. New form of idiopathic osteolysis: nodulosis, arthropathy and osteolysis (NAO) syndrome. Am J Med Genet. 2000;93:5–10. [PubMed: 10861675]
- Azzollini J, Rovina D, Gervasini C, Parenti I, Fratoni A, Cubellis MV, Cerri A, Pietrogrande L, Larizza L. Functional characterisation of a novel mutation affecting the catalytic domain of MMP2 in siblings with multicentric osteolysis, nodulosis and arthropathy. J Hum Genet. 2014;59:631–7. [PubMed: 25273674]
- Bader-Meunier B, Bonafé L, Fraitag S, Breton S, Bodemer C, Baujat G. Mutation in MMP2 gene may result in scleroderma-like skin thickening. Ann Rheum Dis. 2016;75:e1. [PubMed: 26420579]
- Bhavani GS, Shah H, Shukla A, Gupta N, Gowrishankar K, Rao AP, Kabra M, Agarwal M, Ranganath P, Ekbote AV, Phadke SR, Kamath A, Dalal A, Girisha KM. Clinical and mutation profile of multicentric osteolysis nodulosis and arthropathy. Am J Med Genet A. 2016;170A:410–7. [PubMed: 26601801]
- Castberg FC, Kjaergaard S, Mosig RA, Lobl M, Martignetti C, Martignetti JA, Myrup C, Zak M. Multicentric osteolysis with nodulosis and arthropathy (MONA) with cardiac malformation, mimicking polyarticular juvenile idiopathic arthritis: case report and literature review. Eur J Pediatr. 2013;172:1657–63. [PubMed: 23900523]
- de Vos IJHM, Wong ASW, Welting TJM, Coull BJ, van Steensel MAM. Multicentric osteolytic syndromes represent a phenotypic spectrum defined by defective collagen remodeling. Am J Med Genet A. 2019;179:1652–64. [PubMed: 31218820]
- Ekbote AV, Danda S, Zankl A, Mandal K, Maguire T, Ungerer K. Patient with mutation in the matrix metalloproteinase 2 (MMP2) gene - a case report and review of the literature. J Clin Res Pediatr Endocrinol. 2014;6:40–6. [PMC free article: PMC3986738] [PubMed: 24637309]
- Elsebaie H, Mansour MA, Elsayed SM, Mahmoud S, El-Sobky TA. Multicentric Osteolysis, Nodulosis, and Arthropathy in two unrelated children with matrix metalloproteinase 2 variants: Genetic-skeletal correlations. Bone Rep. 2021;15:101106. [PMC free article: PMC8283316] [PubMed: 34307793]
- Evans BR, Mosig RA, Lobl M, Martignetti CR, Camacho C, Grum-Tokars V, Glucksman MJ, Martignetti JA. Mutation of membrane type-1 metalloproteinase, MT1-MMP, causes the multicentric osteolysis and arthritis disease Winchester syndrome. Am J Hum Genet. 2012;91:572–6. [PMC free article: PMC3512002] [PubMed: 22922033]
- Gok F, Crettol LM, Alanay Y, Hacihamdioglu B, Kocaoglu M, Bonafe L, Ozen S. Clinical and radiographic findings in two brothers affected with a novel mutation in matrix metalloproteinase 2 gene. Eur J Pediatr. 2010;169:363–7. [PubMed: 19653001]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Jeong SY, Kim BY, Kim HJ, Yang JA, Kim OH. A novel homozygous MMP2 mutation in a patient with Torg-Winchester syndrome. J Hum Genet. 2010;55:764–6. [PubMed: 20720557]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Kröger L, Löppönen T, Ala-Kokko L, Kröger H, Jauhonen HM, Lehti K, Jääskeläinen J. A novel mutation in the matrix metallopeptidase 2 coding gene associated with intrafamilial variability of multicentric osteolysis, nodulosis, and arthropathy. Mol Genet Genomic Med. 2019;7:e802. [PMC free article: PMC6687624] [PubMed: 31268248]
- Martignetti JA, Aqeel AA, Sewairi WA, Boumah CE, Kambouris M, Mayouf SA, Sheth KV, Eid WA, Dowling O, Harris J, Glucksman MJ, Bahabri S, Meyer BF, Desnick RJ. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat Genet. 2001;28:261–5. [PubMed: 11431697]
- Mosig RA, Dowling O, DiFeo A, Ramirez MC, Parker IC, Abe E, Diouri J, Aqeel AA, Wylie JD, Oblander SA, Madri J, Bianco P, Apte SS, Zaidi M, Doty SB, Majeska RJ, Schaffler MB, Martignetti JA. Loss of MMP-2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum Mol Genet. 2007;16:1113–23. [PMC free article: PMC2576517] [PubMed: 17400654]
- Phadke SR, Ramirez M, Difeo A, Martignetti JA, Girisha KM. Torg-Winchester syndrome: lack of efficacy of pamidronate therapy. Clin Dysmorphol. 2007;16:95–100. [PubMed: 17351352]
- Pichler K, Karall D, Kotzot D, Steichen-Gersdorf E, Rümmele-Waibel A, Mittaz-Crettol L, Wanschitz J, Bonafé L, Maurer K, Superti-Furga A, Scholl-Bürgi S. Bisphosphonates in multicentric osteolysis, nodulosis and arthropathy (MONA) spectrum disorder - an alternative therapeutic approach. Sci Rep. 2016;6:34017. [PMC free article: PMC5043187] [PubMed: 27687687]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Rouzier C, Vanatka R, Bannwarth S, Philip N, Coussement A, Paquis-Flucklinger V, Lambert JC. A novel homozygous MMP2 mutation in a family with Winchester syndrome. Clin Genet. 2006;69:271–6. [PubMed: 16542393]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Temtamy SA, Ismail S, Aglan MS, Ashour AM, Hosny LA, El-Badry TH, Aboul-Ezz EH, Amr K, Fateen E, Maguire T, Ungerer K, Zankl A. A report of three patients with MMP2 associated hereditary osteolysis. Genet Couns. 2012;23:175–84. [PubMed: 22876575]
- Tuysuz B, Mosig R, Altun G, Sancak S, Glucksman MJ, Martignetti JA. A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects. Eur J Hum Genet. 2009;17:565–72. [PMC free article: PMC2721823] [PubMed: 18985071]
- Unger S, Ferreira CR, Mortier GR, Ali H, Bertola DR, Calder A, Cohn DH, Cormier-Daire V, Girisha KM, Hall C, Krakow D, Makitie O, Mundlos S, Nishimura G, Robertson SP, Savarirayan R, Sillence D, Simon M, Sutton VR, Warman ML, Superti-Furga A. Nosology of genetic skeletal disorders: 2023 revision. Am J Med Genet A. 2023. Epub ahead of print. [PMC free article: PMC10081954] [PubMed: 36779427]
- Zankl A, Bonafé L, Calcaterra V, Di Rocco M, Superti-Furga A. Winchester syndrome caused by a homozygous mutation affecting the active site of matrix metalloproteinase 2. Clin Genet. 2005;67:261–6. [PubMed: 15691365]
- Zankl A, Pachman L, Poznanski A, Bonafé L, Wang F, Shusterman Y, Fishman DA, Superti-Furga A. Torg syndrome is caused by inactivating mutations in MMP2 and is allelic to NAO and Winchester syndrome. J Bone Miner Res. 2007;22:329–33. [PubMed: 17059372]
Publication Details
Author Information and Affiliations
Kasturba Medical College of Manipal
Manipal Academy of Higher Education
Manipal, India
Department of Orthopedics
Kasturba Medical College of Manipal
Manipal Academy of Higher Education
Manipal, India
Kasturba Medical College of Manipal
Manipal Academy of Higher Education
Manipal, India
Kasturba Medical College of Manipal
Manipal Academy of Higher Education
Manipal, India
Publication History
Initial Posting: July 14, 2016; Last Revision: March 30, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Bhavani GSL, Shah H, Shukla A, et al. Multicentric Osteolysis Nodulosis and Arthropathy. 2016 Jul 14 [Updated 2023 Mar 30]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.