Summary
Clinical characteristics.
Baller-Gerold syndrome (BGS) can be suspected at birth in an infant with craniosynostosis and upper limb abnormality. The coronal suture is most commonly affected; the metopic, lambdoid, and sagittal sutures may also be involved alone or in combination. Upper limb abnormality can include a combination of thumb hypo- or aplasia and radial hypo- or aplasia and may be asymmetric. Malformation or absence of carpal or metacarpal bones has also been described. Skin lesions may appear anytime within the first few years after birth, typically beginning with erythema of the face and extremities and evolving into poikiloderma. Slow growth is apparent in infancy with eventual height and length typically at 4 SD below the mean.
Diagnosis/testing.
The diagnosis of BGS is established in a proband with typical clinical findings and/or the identification of biallelic pathogenic variants in RECQL4 by molecular genetic testing.
Management.
Treatment of manifestations: Surgery before age six months to repair bilateral craniosynostosis; pollicization of the index finger as needed to create a functional grasp; sunscreen use with poikiloderma to protect against skin cancer.
Surveillance: Because individuals with allelic RECQL4 disorders are at increased risk for osteosarcoma and lymphoma, attention to clinical findings (e.g., bone pain, swelling, and/or limp) for osteosarcoma and lymph node enlargement or generalized symptoms (e.g., fever or unexplained weight loss) for lymphoma is recommended for those with BGS.
Agents/circumstances to avoid: Sun exposure because of risk for skin cancer.
Genetic counseling.
Baller-Gerold syndrome is inherited in an autosomal recessive manner. The parents of an affected child are obligate heterozygotes and therefore carry one pathogenic variant. Heterozygotes (carriers) are asymptomatic. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk family members, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible if both pathogenic variants in the family have been identified.
Diagnosis
Suggestive Findings
Baller-Gerold syndrome should be suspected in individuals with a combination of the following findings:
- Coronal craniosynostosis, manifest clinically as abnormal shape of the skull (brachycephaly) with ocular proptosis and prominent forehead and confirmed by skull x-ray or (preferably) 3D-CT reconstructionWhen the coronal sutures are fused, the orbit is pulled forward. The coronal sutures cannot be discerned on the frontal view, and the same holds true for the lambdoidal sutures.
- Radial ray defect, manifest as aplasia or hypoplasia of the thumb, and/or aplasia or hypoplasia of the radiusNote: Radiographs may be necessary for confirmation of minor radial ray malformations.
- Growth restriction
- Poikiloderma consisting of hyper- and hypopigmentation of the skin with punctate atrophy and telangiectases
Establishing the Diagnosis
The diagnosis of Baller-Gerold syndrome is established in a proband with typical clinical findings and/or by identification of biallelic pathogenic (or likely pathogenic) variants in RECQL4 on molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic RECQL4 variants of uncertain significance (or of one known RECQL4 pathogenic variant and one RECQL4 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of Baller-Gerold syndrome is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with craniosynostosis or those in whom the diagnosis of Baller-Gerold syndrome has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of Baller-Gerold syndrome, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of RECQL4 detects small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected.Perform sequence analysis first. If only one or no pathogenic variant is found, perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.
- A multigene panel that includes RECQL4 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.A multigene panel that also includes deletion/duplication analysis should be considered if only one or no pathogenic variant is found on the multigene panel sequence analysis (see Table 1).
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by craniosynostosis or when the diagnosis of Baller-Gerold syndrome is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
Exome array (when clinically available) may be considered if exome sequencing is not diagnostic.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Baller-Gerold Syndrome
Clinical Characteristics
Clinical Description
Since the original description of Baller-Gerold syndrome (BGS) by Baller [1950] and Gerold [1959], fewer than 40 individuals with BGS have been reported [Mégarbané et al 2000, Van Maldergem et al 2006, Debeljak et al 2009, Siitonen et al 2009, Piard et al 2015, Kaneko et al 2017]. BGS can be suspected at birth in an infant with craniosynostosis and upper limb abnormality. The coronal suture is most commonly affected; the metopic, lambdoid, and sagittal sutures may also be involved alone or in combination [Van Maldergem et al 2016].
Craniofacial findings associated with craniosynostosis
- Brachycephaly
- Proptosis
- Prominent forehead
- Large fontanelles
Additional craniofacial features
- Concave nasal ridge
- Short nose
- Narrow mouth with thin vermilion of the lips
- High arched palate
Skeletal anomalies
- Upper limb anomalies. A combination of thumb hypo- or aplasia and radial hypo- or aplasia is present and may be asymmetric. Malformation or absence of carpal or metacarpal bones has also been described.
- Knee abnormality. Patellar hypo- or aplasia becomes apparent in childhood.
- Late ossification of the patella may be misinterpreted as absence of the patella in infants.
- Absence of patella may result in genu recurvatum and knee instability.
Skin findings. Skin lesions may appear anytime within the first few years after birth.
- Lesions typically begin with erythema of the face and extremities.
- Findings later evolve into poikiloderma (mottled hypo-and hyper-pigmentation, atrophy, and telangiectasias).
Growth. Slow growth is apparent in infancy with eventual height and length typically at 4 SD below the mean.
Development/intelligence. Although intellectual deficiency has been reported [Ramos Fuentes et al 1994], most if not all affected individuals have normal intelligence. No formal studies on intellectual development have been performed.
Other findings
- Imperforate or anterior displacement of the anus has been reported in several individuals.
- Cardiovascular defects such as ventricular septal defects, tetralogy of Fallot, and congenital portal venous malformations have been occasionally described.
Cancer risk. One case of lymphoma was reported in an individual with Baller-Gerold syndrome [Debeljak et al 2009]. However, an increased risk for osteosarcoma, lymphoma, and skin cancer in other disorders associated with pathogenic variants in RECQL4 (see Genetically Related Disorders) has been reported. Therefore, individuals with Baller-Gerold syndrome with symptoms suggestive of cancer should have prompt evaluation.
Genotype-Phenotype Correlations
No formal genotype-phenotype correlations have been made owing to the small number of affected individuals reported to date.
Nomenclature
The name Baller-Gerold syndrome was coined by Cohen [1975] based on descriptions of three affected individuals reported by Baller and Gerold from the German literature.
- Baller [1950] described a woman with short stature, oxycephaly, hypoplasia of the left radius, and aplasia of the right radius; her parents were remotely consanguineous.
- Gerold [1959] described male and female sibs with coronal craniosynostosis, radial and thumb aplasia, and bowing of the ulnae.
Since 1975 the designation Baller-Gerold syndrome has been used to refer to any type of craniosynostosis associated with any type of radial ray defect; this is likely an incorrect use of the term, and has led some authors to consider metopic ridging and radial ray defects observed in valproate embryopathy sufficient for a diagnosis of BGS [Santos de Oliveira et al 2006].
Prevalence
The prevalence of Baller-Gerold syndrome is unknown; it is probably less than 1:1,000,000 [Mo et al 2018].
Genetically Related (Allelic) Disorders
Pathogenic variants in RECQL4 have also been identified in individuals with Rothmund-Thomson syndrome [Kitao et al 1999] and RAPADILINO syndrome [Siitonen et al 2003].
- Rothmund-Thomson syndrome (RTS) is characterized by poikiloderma; sparse hair, eyelashes, and/or eyebrows; small stature; skeletal and dental abnormalities; cataracts; and an increased risk for cancer, especially osteosarcoma. The skin is typically normal at birth; the rash of RTS develops between ages three and six months as erythema, swelling, and blistering on the face and subsequently spreads to the buttocks and extremities. The rash evolves over months to years into the chronic pattern of reticulated hypo- and hyperpigmentation, punctate atrophy, and telangiectasias, collectively known as poikiloderma. Hyperkeratotic lesions occur in approximately one third of individuals. Skeletal abnormalities include radial ray defects, ulnar defects, absent or hypoplastic patella, and osteopenia.The diagnosis of RTS is established by clinical findings. Molecular testing is confirmatory and may be useful in situations in which clinical findings are atypical. Identification of biallelic pathogenic variants in RECQL4 on molecular genetic testing establishes the diagnosis if clinical features are inconclusive.
- RAPADILINO syndrome (OMIM 266280) is an acronym for radial ray defect; patellae hypoplasia or aplasia and cleft or highly arched palate; diarrhea and dislocated joints; little size and limb malformation; nose slender and normal intelligence. It is characterized by pre- and postnatal growth retardation. Cervical spine segmentation defects have been reported. Failure to thrive results from feeding problems and juvenile diarrhea of unknown cause [Siitonen et al 2003]. Since its original description in Finland [Kääriäinen et al 1989], only 14 Finnish and two non-Finnish individuals have been reported [Vargas et al 1992, Kant et al 1998, Jam et al 1999, Siitonen et al 2003]. Osteosarcoma was reported in one of the 16 individuals. Lymphoma appears to be a frequent complication in individuals with RAPADILINO; it occurred in four affected individuals before age 35 years [Siitonen et al 2009].The Finn-specific RECQL4 splice site variant (IVS7+2delT) associated with RAPADILINO syndrome leads to in-frame skipping of exon 7 that is predicted to remove 44 amino acids just before the conserved helicase domain, apparently without altering transcription of the helicase domain itself. Nine of the 14 affected Finnish individuals are homozygous for IVS7+2delT and five are compound heterozygotes for IVS7+2delT and a nonsense variant in extra-helicase exons 5, 18, and 19, thus sparing in all cases the helicase domain, which is therefore thought to play a role in poikiloderma and predisposition to osteosarcoma [Siitonen et al 2003].
RTS, RAPADILINO syndrome, and BGS share the clinical features of pre- and postnatal growth retardation, chronic diarrhea, and patellar hypo- or aplasia. Radial hypo- or aplasia is almost always present in individuals with RAPADILINO syndrome and BGS and less frequently seen in those with RTS. Poikiloderma, a characteristic of RTS and also described in BGS, is not seen in RAPADILINO syndrome. However, the absence of poikiloderma cannot be confirmed before age one year because of its late onset. Craniosynostosis, particularly affecting the coronal suture, is a diagnostic feature of BGS, and is rarely seen in RTS. Alopecia and absence of eyelashes and brows, characteristics of RTS, have not been observed in individuals with BGS or RAPADILINO.
Differential Diagnosis
The major differential diagnosis for Baller-Gerold syndrome (BGS) comprises the allelic disorders Rothmund-Thomson syndrome and RAPADILINO syndrome (OMIM 266280). (See Genetically Related Disorders.) See Figure 1.
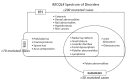
Figure 1.
Diagram showing overlapping and unique clinical features of the RECQL4-associated disorders. Note: "Mutated cases" refers to cases with a molecular diagnosis. From Van Maldergem et al [2016]
Additional conditions to consider are included in Table 2.
Table 2.
Disorders to Consider in the Differential Diagnosis of BGS
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease in an individual diagnosed with Baller-Gerold syndrome (BGS), the following are recommended if they have not already been completed:
- Consultation with a clinical geneticist and/or genetic counselor
- Neurosurgery or craniofacial specialist consultation for evaluation of craniosynostosis
- Orthopedic surgery and occupational therapy assessment to evaluate hand and arm function and need for surgery
- Dermatology evaluation if poikiloderma develops
Treatment of Manifestations
Craniosynostosis should be managed by neurosurgical/craniofacial specialists. When craniosynostosis is bilateral, surgery is usually performed before age six months.
Pollicization of the index finger to restore a functional grasp has had satisfactory results in a number of persons with absence of the thumb [Foucher et al 2005]. However, many children with aplasia of the thumb are able to function without orthopedic surgical intervention.
If poikiloderma is present, sensible use of sunscreens may protect against potential risk for skin cancer due to UV exposure.
If cancer arises, medical care should be sought from an oncologist familiar with the type of cancer.
Surveillance
Although lymphoma has only been described in one individual with BGS to date [Debeljak et al 2009], it is known that individuals with RECQL4 pathogenic variants associated with both Rothmund-Thomson syndrome and RAPADILINO syndrome are at increased risk for developing osteosarcoma and lymphoma. Given the potential risk, it would be reasonable for affected individuals with BGS and RECQL4 pathogenic variants (or their guardians) to be aware of the signs and symptoms associated with these malignancies. These signs and symptoms may include bone pain, swelling, and/or limp for osteosarcoma, and lymph node enlargement or generalized symptoms such as fever or unexplained weight loss for lymphoma.
Agents/Circumstances to Avoid
Excessive sun exposure should be avoided because of the theoretic increased risk for skin cancer.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Baller-Gerold syndrome (BGS) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers of one pathogenic variant).
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. The offspring of an individual with BGS are obligate heterozygotes (carriers) for a pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the RECQL4 pathogenic variants in the family.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring, and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the RECQL4 pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Ultrasound examination. Serial ultrasound examination may identify limb shortening, radial hypo/aplasia, and abnormal head shape (brachycephaly). Ultrasound examination revealing these findings from 14 weeks' gestation onward identified BGS in at-risk pregnancies [Van Maldergem et al 1992, Siitonen et al 2009, Cao et al 2015].
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Children's Craniofacial AssociationPhone: 800-535-3643Email: contactCCA@ccakids.com
- FACES: National Craniofacial AssociationPhone: 800-332-2373; 423-266-1632Email: info@faces-cranio.org
- National Institute of Neurological Disorders and Stroke (NINDS)Phone: 800-352-9424
- REACHHelping children with upper limb differences live life without limits.United KingdomPhone: 0845 1306 225; 020 3478 0100
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Baller-Gerold Syndrome: Genes and Databases
Table B.
OMIM Entries for Baller-Gerold Syndrome (View All in OMIM)
Molecular Pathogenesis
RECQL4, pathogenic variants in which cause BGS, encodes ATP-dependent DNA helicase Q4, a protein of 1,208 amino acids belonging to superfamily II of helicases, known as RecQ, categorized by a 3'-5' polarity of unwinding double-stranded DNA and RNA-DNA hybrids to produce single-stranded DNA templates. At least five RecQ human paralogs are known with distinct but partially overlapping roles: RECQL, BLM, WRN, RECQL4, and RECQL5. Pathogenic variants in BLM are associated with Bloom syndrome; pathogenic variants in WRN are associated with Werner syndrome; both are autosomal recessive chromosome instability conditions characterized by predisposition to cancer and/or premature aging. These features are shared by RECQL4-associated autosomal recessive RTS and by the phenotypically overlapping RAPADILINO and BGS, characterized by genetic instability, growth deficiency, and cancer predisposition. To date, no human disease has been associated with RECQL or RECQL5.
RecQ helicases have essential functions not only at various stages of DNA processing (replication, recombination, repair, telomere maintenance) but also in translation, RNA processing, mtDNA maintenance, and chromosome segregation [Croteau et al 2014]. Since they act in virtually all aspects of DNA metabolism, perturbation of their expression and biochemical activity leads to genomic instability, resulting in disease and cancer predisposition [Bochman 2014].
Gene structure. RECQL4 has 21 exons, spanning more than 6.5 kb. The gene has a coding sequence consisting of 3,627 bp, and its expression is regulated by a housekeeping promoter containing the binding sites for the transcription factors Sp1 and AP2. It encodes a 133-kd protein of 1,208 amino acids, RECQ protein-like 4 (RECQL4), which contains the DNA helicase domain exons homologous to the E coli RecQ helicase and shared by all five members of the RecQ family in humans. The RECQL4 helicase domain, including the ATP binding domain (aa 489 to 662) and the C-terminal domain (aa 683 to 850) is encoded by exons 8 to 15. RECQL4 is unique for having 13 introns composed of fewer than 100 bp, a feature predisposing to inefficient splicing [Wang et al 2002]. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. To date, 11 different pathogenic variants have been identified in seven families:
- c.3056-2A>C (homozygous) [Mégarbané et al 2000, Van Maldergem et al 2006]
- c.3061C>T (p.Arg1021Trp) and c.1573del – compound heterozygous in two families [Van Maldergem et al 1992, Van Maldergem et al 2006, Siitonen et al 2009]
- c.2335_2356del (homozygous) [Siitonen et al 2009]
- c.496C>T (p.Gln166Ter) and c.3151A>G (p.Ile1051Val) – compound heterozygous in two terminated pregnancies [Siitonen et al 2009]
- c.2492_2493del and c.2506_2518del – compound heterozygous in a child with a severe BGS phenotype who developed a midline NK/T lymphoma at age 2.5 years [Debeljak et al 2009]
- c.2059-1G>C and c.2141_2142del – compound heterozygous in a fetus with (prenatally diagnosed) severe BGS [Cao et al 2015]
- An intron 12 through exon 18 deletion (1,614-bp deletion and 1-bp G insertion; g.145737562_145739175delinsC) – homozygous in a Japanese boy age four years with café au lait-like spots but no poikiloderma [Kaneko et al 2017]; predicted to remove the helicase motif from IV to VI
Interestingly both missense variants p.Arg1021Trp and p.Ile1051Val are located in the RecQ C-terminal region (RQC), which is essential for proper function not only of RECQL4 but also of its paralogs [Mojumdar et al 2017].
Of the 11 RECQL4 pathogenic variants reported to date:
- Seven have been detected only in BGS.
- The p.Arg1021Trp and c.2492_2493del variants were observed in BGS and RTS.
- The recurrent c.1573del has been observed in BGS, RTS, and RAPADILINO.
- The c.2059-1G>C variant was detected in a fetus with BGS and was reported in one individual with RTS [Beghini et al 2003]. Of note, other base changes at the same site were observed in either RTS (c.2059-1G>T) [Kitao et al 1999, Kellermayer et al 2005, Siitonen et al 2009, Piard et al 2015] or RAPADILINO (c.2059-1G>A) [Siitonen et al 2009].
Notwithstanding the small number of unrelated cases confirmed by molecular diagnosis, compound heterozygosity is observed in more than half of the cases (4 vs 3 homozygotes), in line with the trend of RTS, where about two thirds of affected individuals reported are compound heterozygotes [Van Maldergem et al 2016].
Table 3.
RECQL4 Pathogenic Variants Discussed in This GeneReview
Normal gene product. Processing of aberrant DNA structures that arise during DNA replication and repair appears to be a major function of ATP-dependent DNA helicase Q4, the protein encoded by RECQL4 . Disruption of DNA replication fork progression by damage-induced stable secondary structures can impede replication fork progression, resulting in arrest or collapse of the fork leading through impairment in the removal of these aberrant structures to chromosome instability and, ultimately, cell death or cancer. In this respect, ATP-dependent DNA helicase Q4 can be considered a caretaker of the genome [Wu & Hickson 2006].
RECQL4 is a highly multifunctional protein whose biologic role is partially interconnected with the other RecQ family proteins. Its helicase activity, questioned for some time, has been fully demonstrated and assigned to RECQL4 conserved helicase motif and N-terminal domain between amino acids 240 and 400, each independently promoting ATP-dependent DNA unwinding [Xu & Liu 2009]. Also, the puzzling lack of a physical RecQ C-terminal region (RQC), which is an essential part of the catalytic core of all other human paralogs, has now been resolved by the experimental demonstration of the presence of a functional RQC domain in human RECQL4 [Mojumdar et al 2017]. Like all human RecQs, RECQL4 is essential for DNA recombination and DNA repair [Croteau et al 2014] and is involved in telomere maintenance in concert with BLM and WRN [Ghosh et al 2012]. As regards DNA replication, RECQL and RECQL4 are integral components of the human replication complex and play distinct roles in DNA replication initiation and replication fork progression [Xu & Liu 2009, Thangavel et al 2010]. It has been proposed that the helicase activity of RECQL4 may be specialized in the rescue of stalled replication forks or may act in the initial unwinding at the replication origin [Masai 2011].
Absolutely unique to RECQL4 is regulation of maintenance of mitochondrial DNA copy number and transport of p53 (pathogenic variants in which cause Li-Fraumeni syndrome) to mitochondria [De et al 2012]. Both RECQL4 and p53 potentiate the activity of polymerase γ, maintaining the integrity of the human mitochondrial genome [Gupta et al 2014]. Last, but of foremost importance for cancer predisposition associated to defective RECQL4, RECQL4 has an essential role in ensuring correct chromosome segregation through its stable interaction with the ubiquitin ligases UBR1 and UBR2 [Yin et al 2004] involved in the N-end-rule pathway, essential for chromosome stability [Rao et al 2001]. The interconnection between RECQL4 and cohesin pathway is evidenced by downregulation of RECQL4 in cells from individuals with Cornelia de Lange syndrome [Liu et al 2009] and inclusion of RECQL4 among the accessory proteins acting in the cohesin pathway.
Abnormal gene product. Most of the identified RECQL4 variants are frameshift or nonsense variants, predicted to destabilize the mature mRNA and to result in low levels of truncated proteins [Ouyang et al 2008]. Maintenance of the N-terminus, which is essential in initiation of DNA replication, appears indispensable for cell viability, consistent with the finding of rare pathogenic variants (not in homozygous state) in this region in individuals with all RECQL4-related syndromes. The as-yet-limited functional studies of mutated RECQL4 proteins lend support for a helicase-dependent cellular function of RECQL4 [Croteau et al 2012], confirming the postulated association between deleterious variants affecting helicase activity and osteosarcoma [Wang et al 2003].
Chapter Notes
Revision History
- 19 April 2018 (ha) Comprehensive update posted live
- 7 June 2011 (me) Comprehensive update posted live
- 13 August 2007 (me) Review posted live
- 23 April 2007 (lvm) Original submission
References
Literature Cited
- Baller F. Radiusaplasie und Inzucht. Z Menschl Vererb Konstitutionsl. 1950;29:782–90.
- Beghini A, Castorina P, Roversi G, Modiano P, Larizza L. RNA processing defects of the helicase gene RECQL4 in a compound heterozygous Rothmund-Thomson patient. Am J Med Genet A. 2003;120A:395–9. [PubMed: 12838562]
- Bochman ML. Roles of DNA helicases in the maintenance of genome integrity. Mol Cell Oncol. 2014;1:e963429. [PMC free article: PMC4905024] [PubMed: 27308340]
- Cao DH, Mu K, Liu DN, Sun JL, Bai XZ, Zhang N, Qiu GB, Ma XW. Identification of novel compound heterozygous RECQL4 mutations and prenatal diagnosis of Baller-Gerold syndrome: a case report. Genet Mol Res. 2015;14:4757–66. [PubMed: 25966250]
- Cohen MM Jr. An etiologic and nosologic overview of craniosynostosis syndromes. Birth Defects Orig Artic Ser. 1975;11:137–89. [PubMed: 179637]
- Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem. 2014;83:519–52. [PMC free article: PMC4586249] [PubMed: 24606147]
- Croteau DL, Rossi ML, Ross J, Dawut L, Dunn C, Kulikowicz T, Bohr VA. RAPADILINO RECQL4 mutant protein lacks helicase and ATPase activity. Biochim Biophys Acta. 2012;1822:1727–34. [PMC free article: PMC3500628] [PubMed: 22885111]
- De S, Kumari J, Mugdal R, Modi P, Gupta S, Futami K, Goto H, Lindor NM, Furuichi Y, Mohanty D, Sengupta S. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress. J Cell Sci. 2012;125:2509–22. [PubMed: 22357944]
- Debeljak M, Zver A, Jazbec J. A patient with Baller-Gerold syndrome and midline NK/T lymphoma. Am J Med Genet A. 2009;149A:755–9. [PubMed: 19291770]
- Foucher G, Medina J, Lorea P, Pivato G. Principalization of pollicization of the index finger in congenital absence of the thumb. Tech Hand Up Extrem Surg. 2005;9:96–104. [PubMed: 16201251]
- Gerold M. Healing of a fracture in an unusual case of congenital anomaly of the upper extremities. Zentralbl Chir. 1959;84:831–4. [PubMed: 13669699]
- Ghosh AK, Rossi ML, Singh DK, Dunn C, Ramamoorthy M, Croteau DL, Liu Y, Bohr VA. RECQL4, the protein mutated in Rothmund-Thomson syndrome, functions in telomere maintenance. J Biol Chem. 2012;287:196–209. [PMC free article: PMC3249070] [PubMed: 22039056]
- Gupta S, De S, Srivastava V, Hussain M, Kumari J, Muniyappa K, Sengupta S. RECQL4 and p53 potentiate the activity of polymerase γ and maintain the integrity of the human mitochondrial genome. Carcinogenesis. 2014;35:34–45. [PubMed: 24067899]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Jam K, Fox M, Crandall BF. RAPADILINO syndrome: a multiple malformation syndrome with radial and patellar aplasia. Teratology. 1999;60:37–8. [PubMed: 10413338]
- Kääriäinen H, Ryöppy S, Norio R. RAPADILINO syndrome with radial and patellar aplasia/hypoplasia as main manifestations. Am J Med Genet. 1989;33:346–51. [PubMed: 2801769]
- Kaneko H, Izumi R, Oda H, Ohara O, Sameshima K, Ohnishi H, Fukao T, Funato M. Nationwide survey of Baller-Gerold syndrome in Japanese population. Mol Med Rep. 2017;15:3222–4. [PubMed: 28358413]
- Kant SG, Baraitser M, Milla PJ, Winter RM. Rapadilino syndrome--a non-Finnish case. Clin Dysmorphol. 1998;7:135–8. [PubMed: 9571286]
- Kellermayer R, Siitonen HA, Hadzsiev K, Kestila M, Kosztolanyi G. A patient with Rothmund-Thomson syndrome and all features of RAPADILINO. Arch Dermatol. 2005;141:617–20. [PubMed: 15897384]
- Kitao S, Shimamoto A, Goto M, Miller RW, Smithson WA, Lindor NM, Furuichi Y. Mutations in RECQL4 cause a subset of cases of Rothmund-Thomson syndrome. Nat Genet. 1999;22:82–4. [PubMed: 10319867]
- Klopocki E, Schulze H, Strauss G, Ott CE, Hall J, Trotier F, Fleischhauer S, Greenhalgh L, Newbury-Ecob RA, Neumann LM, Habenicht R, Konig R, Seemanova E, Mégarbané A, Ropers HH, Ullmann R, Horn D, Mundlos S. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am J Hum Genet. 2007;80:232–40. [PMC free article: PMC1785342] [PubMed: 17236129]
- Larizza L, Roversi G, Verloes A. Clinical utility gene card for: Rothmund-Thomson syndrome. Eur J Hum Genet. 2013;21(7) [PMC free article: PMC3722954] [PubMed: 23188052]
- Liu J, Zhang Z, Bando M, Itoh T, Deardorff MA, Clark D, Kaur M, Tandy S, Kondoh T, Rappaport E, Spinner NB, Vega H, Jackson LG, Shirahige K, Krantz ID. Transcriptional dysregulation in NIPBL and cohesin mutant human cells. Plos Biol. 2009;7:e1000119. [PMC free article: PMC2680332] [PubMed: 19468298]
- Masai H. RecQL4: a helicase linking formation and maintenance of a replication fork. J Biochem. 2011;149:629–631. [PubMed: 21436139]
- Mégarbané A, Melki I, Souraty N, Gerbaka J, El Ghouzzi V, Bonaventure J, Mornand A, Loiselet J. Overlap between Baller-Gerold and Rothmund-Thomson syndrome. Clin Dysmorphol. 2000;9:303–5. [PubMed: 11045594]
- Mendoza-Londono R, Lammer E, Watson R, Harper J, Hatamochi A, Hatamochi-Hayashi S, Napierala D, Hermanns P, Collins S, Roa BB, Hedge MR, Wakui K, Nguyen D, Stockton DW, Lee B. Characterization of a new syndrome that associates craniosynostosis, delayed fontanel closure, parietal foramina, imperforate anus, and skin eruption: CDAGS. Am J Hum Genet. 2005;77:161–8. [PMC free article: PMC1226190] [PubMed: 15924278]
- Mo D, Zhao Y, Balajee AS. Human RecQL4 helicase plays multifaceted roles in the genomic stability of normal and cancer cells. Cancer Lett. 2018;413:1–10. [PubMed: 29080750]
- Mojumdar A, De March M, Marino F, Onesti S. The human RecQ4 helicase contains a functional RecQ C-terminal region (RQC) that is essential for activity. J Biol Chem. 2017;292:4176–84. [PMC free article: PMC5354486] [PubMed: 27998982]
- Ouyang KJ, Woo LL, Ellis NA. Homologous recombination and maintenance of genome integrity: cancer and aging through the prism of human RecQ helicases. Mech Aging Dev. 2008;129:425–40. [PubMed: 18430459]
- Piard J, Aral B, Vabres P, Holder-Espinasse M, Mégarbané A, Gauthier S, Capra V, Pierquin G, Callier P, Baumann C, Pasquier L, Baujat G, Martorell L, Rodriguez A, Brady AF, Boralevi F, González-Enseñat MA, Rio M, Bodemer C, Philip N, Cordier MP, Goldenberg A, Demeer B, Wright M, Blair E, Puzenat E, Parent P, Sznajer Y, Francannet C, DiDonato N, Boute O, Barlogis V, Moldovan O, Bessis D, Coubes C, Tardieu M, Cormier-Daire V, Sousa AB, Franques J, Toutain A, Tajir M, Elalaoui SC, Geneviève D, Thevenon J, Courcet JB, Rivière JB, Collet C, Gigot N, Faivre L, Thauvin-Robinet C. Search for ReCQL4 mutations in 39 patients genotyped for suspected Rothmund-Thomson/Baller-Gerold syndromes. Clin Genet. 2015;87:244–51. [PubMed: 24635570]
- Ramos Fuentes FJ, Nicholson L, Scott CI Jr. Phenotypic variability in the Baller-Gerold syndrome: report of a mildly affected patient and review of the literature. Eur J Pediatr. 1994;153:483–7. [PubMed: 7957363]
- Rao H, Uhlmann F, Nasmith K, Varshavsky A. Degradation of a cohesion subunit by the N-end rule pathway is essential for chromosomal stability. Nature. 2001;410:955–9. [PubMed: 11309624]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Santos de Oliveira R, Lajeunie E, Arnaud E, Renier D. Fetal exposure to sodium valproate associated with Baller-Gerold syndrome: case report and review of the literature. Childs Nerv Syst. 2006;22:90–4. [PubMed: 15789214]
- Siitonen HA, Kopra O, Kaariainen H, Haravuori H, Winter RM, Saamanen AM, Peltonen L, Kestila M. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum Mol Genet. 2003;12:2837–44. [PubMed: 12952869]
- Siitonen HA, Sotkasiira J, Biervliet M, Benmansour A, Capri Y, Cormier-Daire V, Crandall B, Hannula-Jouppi K, Hennekam R, Herzog D, Keymolen K, Lipsanen-Nyman M, Miny P, Plon SE, Riedl S, Sarkar A, Vargas FR, Verloes A, Wang LL, Kääriäinen H, Kestilä M. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. 2009;17:151–8. [PMC free article: PMC2986053] [PubMed: 18716613]
- Thangavel S, Mendoza-Maldonado R, Tissino E, et al. Human RECQ1 and RECQL4 helicases play distinct roles in DNA replication initiation. Mol Cell Biol. 2010;30:1382–1396. [PMC free article: PMC2832491] [PubMed: 20065033]
- Van Maldergem L, Piard J, Larizza L, Wang LL. RECQL4-related phenotypes. In: Erickson RP, Wynshaw-Boris AJ, eds. Epstein's Inborn Errors of Development: The Molecular Basis of Clinical Disorders. 3 ed. New York, NY: Oxford University Press; 2016.
- Van Maldergem L, Siitonen HA, Jalkh N, Chouery E, De Roy M, Delague V, Muenke M, Jabs EW, Cai J, Wang LL, Plon SE, Fourneau C, Kestila M, Gillerot Y, Mégarbané A, Verloes A. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J Med Genet. 2006;43:148–52. [PMC free article: PMC2564634] [PubMed: 15964893]
- Van Maldergem L, Verloes A, Lejeune L, Gillerot Y. The Baller-Gerold syndrome. J Med Genet. 1992;29:266–8. [PMC free article: PMC1015930] [PubMed: 1583650]
- Vargas FR, de Almeida JC, Llerena JC Junior, Reis DF. RAPADILINO syndrome. Am J Med Genet. 1992;44:716–9. [PubMed: 1481838]
- Wang LL, Gannavarapu A, Kozinetz CA, Levy ML, Lewis RA, Chintagumpala MM, Ruiz-Maldanado R, Contreras-Ruiz J, Cunniff C, Erickson RP, Lev D, Rogers M, Zackai EH, Plon SE. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst. 2003;95:669–74. [PubMed: 12734318]
- Wang LL, Worley K, Gannavarapu A, Chintagumpala MM, Levy ML, Plon SE. Intron-size constraint as a mutational mechanism in Rothmund-Thomson syndrome. Am J Hum Genet. 2002;71:165–7. [PMC free article: PMC384974] [PubMed: 12016592]
- Wu L, Hickson ID. DNA helicases required for homologous recombination and repair of damaged replication forks. Annu Rev Genet. 2006;40:279–306. [PubMed: 16856806]
- Xu X, Liu Y. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein. Embo J. 2009;28:568–77. [PMC free article: PMC2657580] [PubMed: 19177149]
- Yin J, Kwon YT, Varshavsky A, Wang W. RECQL4, mutated in the Rothmund-Thomson and RAPADILINO syndromes, interacts with ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Hum Mol Genet. 2004;13:2421–30. [PubMed: 15317757]
Publication Details
Author Information and Affiliations
CHRU de Besançon – Hôpital Saint-Jacques
Besançon, France
IRCSS Istituto Auxologico Italiano
Milan, Italy
Department of Pediatrics
Texas Children's Hospital
Baylor College of Medicine
Houston, Texas
Publication History
Initial Posting: August 13, 2007; Last Update: April 19, 2018.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Van Maldergem L, Piard J, Larizza L, et al. Baller-Gerold Syndrome. 2007 Aug 13 [Updated 2018 Apr 19]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
