Abstract
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disorder, characterized by the degeneration of upper and lower motor neurons of the motor cortex, brainstem, and ventral horn of the spinal cord. The role of glial cells in the onset and progression of ALS is increasingly being recognized. Dysfunctional astrocytes, with an atypical and neurotoxic phenotype, in the cerebral cortex and the spinal cord promote neuroinflammation and motor neuron degeneration. Indeed, cortical and spinal cord astrocytes from SOD1G93A (mSOD1) mice are neurotoxic, develop early deficits, and lose their neuro-supportive properties before disease onset. This chapter discusses the contribution of dysfunctional cortical and spinal cord astrocytes in the development and progression of ALS. Differences in astrocyte heterogeneity and reactivity, calcium signaling, neurotransmitters, and in paracrine signaling mechanisms along with implications for novel therapies in ALS are addressed.
Keywords:
amyotrophic lateral sclerosis, astrocyte subpopulations, glutamate and homeostatic imbalance, reactive biomarkers, revival of dysfunctional astrocytesINTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a progressive and devastating neurodegenerative disorder, characterized by the degeneration of upper and lower motor neurons (MNs) across the corticospinal tract, from the motor cortex to the brainstem, and ventral horn of the spinal cord (SC) (1). The disease progression is aggressive, with a fatal outcome usually within five years of onset. Currently, there is no cure and very few treatments are available for this devastating MN disease. While most cases of ALS are sporadic (sALS) (90–95%), a small subset (5–10%) of patients have a positive familial history (fALS) (2). Mutations in the gene encoding for the Cu2+/Zn2+ ion-binding superoxide dismutase (SOD1) protein are the most common and represent approximately 20% of fALS cases. SOD1G93A mouse (mSOD1) is currently the most widely used animal model to study ALS. Other mutations associated to ALS are TARDBP (also known as TDP-43; encodes for TAR DNA-binding protein), FUS (encodes RNA-binding protein Fused in Sarcoma/Translocated in Sarcoma), ANG (encodes angiogenin, ribonuclease, RNase A family, 5), and OPTN (encodes optineurin) (2). The most common mutations associated to ALS and frontotemporal dementia, a variant of ALS, are the gain of toxicity by the nucleotide GGGGCC repeat expansions within the gene C9ORF72 (3).
The relevance of glial cells on the onset and progression of ALS is now recognized. In genetically modified mice, in which the SOD1 mutation was selectively excised from different central nervous system (CNS) cell types, it was observed that different glial cells significantly promote disease progression (4). Among these, dysfunctional astrocytes, with an aberrant and neurotoxic phenotype in the cerebral cortex and the SC of mSOD1 mice, were recognized as major contributors. ALS astrocytes develop early deficits and lose neuro-supportive properties, secreting toxic factors that directly induce MN cell death (5, 6). In this chapter, we discuss the role of dysfunctional astrocytes in ALS with emphasis on astrocyte reactivity and heterogeneity, neurotransmitter transporters, and dysregulation of autocrine and paracrine mechanisms.
ASTROCYTIC REACTIVITY AND HETEROGENEITY
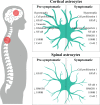
The exact mechanisms for neuronal degeneration in ALS are still unclear, but astrocytes are recognized as important players in both upper and lower MN loss (4). Neuroinflammation and glial activation are observed at the onset of, and during, disease progression. Astrocytes express differential astrocytic receptors, transporters, and neurotransmitters, and release neurotrophic factors, inflammatory mediators and cytotoxins. These reactive astrocytes are observed in the cortex and SC of ALS patients, both in sALS and fALS cases (7). Glial cell proliferation and activation are found not only in motor areas, but also in non-motor areas, such as hippocampus, of mSOD1 rats, starting at the presymptomatic stage of the disease (8). SC mSOD1 astrocytes from newborn pups were shown to cause MN toxicity, long before any visible reactive gliosis (5). In ALS, these reactive astrocytes lose their physiological and homeostatic functions and acquire a neurotoxic and aberrant phenotype (5). Transplantation of SOD1 glial-restricted precursor cells into the SC of healthy rodents showed to differentiate into neurotoxic astrocytes and trigger MN degeneration (9), whereas transplantation of normal astrocyte precursors delayed disease progression and extended the survival of mSOD1 rats (10). Astrocytes derived from sALS patients also led to MN degeneration after transplantation into mice (11). Thus, the identification of specific mechanisms and mediators of astrocyte toxicity offers important insights into the pathways of MN degeneration in ALS and the ways to prevent them. Both upper and lower MNs are affected in ALS, and astrocytes reveal regional diverse phenotypes, as depicted in .
Astrocyte regional diversity and heterogeneity in ALS. Astrocytes in ALS are known to have an aberrant and reactive profile, expressing different astrocytic markers depending on their location in the central nervous system (cortical or spinal astrocytes), (more...)
Cortical and SC astrocytes cause neuronal dysfunction by specific and common pathological mechanisms (12). This is in line with the recent concept of astrocyte heterogeneity, either in the same zone of the CNS or across different regions (13). Reactive astrogliosis and graded reactions depend on microenvironmental cues and interactions between neighboring cells, as well as on autocrine signaling (14). In the mSOD1 mouse model, cortical astrocytes present an early hypertrophic/fibroblast-like morphology and a reactive and inflammatory phenotype. Such phenotype is characterized by decreased expression of glial fibrillary acidic protein (GFAP) and increased cell proliferative capacity, as well as elevated expression of S100 calcium (Ca2+)-binding protein B (S100B) and high mobility group box protein 1 (HMGB1). SC astrocytes appear to be more constrained than cortical ones, mainly in the presymptomatic stage, with decreased S100B and HMGB1 expression levels (12, 15). In late stages, a marked proliferative capacity and overexpression of S100B and HMGB1 is observed in astrocytes from the SC of ALS patients, and rodent models (16, 17).
GFAP is the hallmark intermediate filament protein in astrocytes and its upregulation is usually associated with reactive astrogliosis (18). However, GFAP amount in adjacent astrocytes is extremely heterogeneous, as well as its expression in different regions (19, 20). Reactive GFAP-astrocytes were found in the ventral horn of ALS patients (21), with elevated appearance in the cerebrospinal fluid (CSF) relative to other neurologic diseases (22). Astrocytes with increased GFAP content and multiple inflammatory/reactive mediators were also identified in the SC of adult mice (23). While differential distribution of GFAP immunoreactivity was found in the white matter of the SC at early symptomatic transgenic mSOD1 mice, in the gray matter that was found only in the end-stage disease (24). In other studies, GFAP expression did not show differences between mSOD1 and wild-type mice at 40 and 80 postnatal days, but strongly increased at terminal stages in the SC of mSOD1 mice (25). In contrast, our studies evidenced decreased levels of GFAP in the pre-symptomatic stage in the cortical brain (15) and in the SC of mSOD1 mice (17). Decreased GFAP expression levels were also found in astrocytes isolated from the brain cortex of mSOD1 pups, which presented aberrant astrocytic markers [increased S100B, connexin 43 (Cx43), Ki-67, and vimentin, together with decreased GFAP, glutamate transporter-1 (GLT-1) and glutamate/aspartate transporter (GLAST)], as found in the same region at the symptomatic stage (15). Such acquired “immature” or dedifferentiated astrocyte phenotype is neurotoxic and disease-specific in the cortical brain, and probably associated with the bulbar origin of the disorder (12). The early occurrence of such signature in 7-day-old astrocytes from the brain cortex of mSOD1 mice was also observed in other disease models and related with the loss of neuro-supportive functions (26). Decreased expression of GFAP was similarly found in glial cell populations from the SC of symptomatic mSOD1 rats (16). When expression of mSOD1 was virally induced in cortical astrocytes, alterations in cell morphology and density, together with low GFAP immunostaining were obtained (27). However, the loss of GFAP only marginally accelerated disease progression in the SOD1H46R transgenic mice (28). In sum, GFAP is not an absolute marker of reactivity, nor it strictly correlates with the disease severity. Variations in animal models, regional diversity, and specific astrocyte subpopulations may be the reason for the disparate data found in the literature.
Increased expression of HMGB1 in reactive glia may lead to the activation of toll-like receptor/receptor for advanced glycation end-products (TLR/RAGE) signaling pathways, and contribute to the progression of inflammation and MN injury (29). S100B is a Ca2+-binding protein that is highly expressed in astrocytes and, depending on its concentration, can have beneficial or deleterious effects. In ALS, S100B levels are increased in the CSF, positively correlating with a worse prognosis of the disease (30). The inhibition of S100B downregulates the expression of GFAP and cytokines, such as tumor necrosis factor (TNF)-α, C-C motif chemokine ligand 6 (CCL6), and C-X-C motif chemokine ligand 10 (CXCL10), indicating its association to a proinflammatory phenotype in mSOD1 astrocytes. Expression and release of pro-inflammatory cytokines lead to the activation of the nuclear factor kappa B (NF-κB) signaling cascade, a regulator of reactive gliosis and inflammation. In early stages of ALS, NF-κB activation in SC astrocytes can induce a neuroprotective phenotype, by promoting beneficial microglia activation and delaying disease progression. However, prolonged NF-κB activation in later stages exacerbates the immune response with pro-inflammatory microglial activation, gliosis, and disruption of the blood-SC barrier (31). Communication among astrocytes is promoted by connexin-based gap junctions, such as Cx43. Abnormally high Cx43 expression in the cortical and SC astrocytes of mSOD1 mice and ALS patients is associated to astrocyte-mediated neurotoxicity (32).
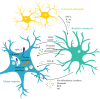
Astrocytes in ALS release soluble toxic factors that promote MN degeneration (16), but still not clearly identified. Upregulation of the major histocompatibility complex I (MHC-I) in MNs seems to be associated with a slower disease progression (4). However, astrocytes in both mSOD1 mice and ALS patients were shown to downregulate the expression of MHC-I in MNs, by causing endoplasmic reticulum (ER) stress, thus increasing their susceptibility to astrocyte-induced cell death (33). On the other hand, reactive astrocytes secrete increased transforming growth factor (TGF)-β1 that causes MN autophagic dysregulation, abnormal protein aggregation, and cellular toxicity (23). Astrocytes, when exposed to the CSF from ALS patients, release pro-inflammatory cytokines, such as interleukin (IL)-6, TNF-α, and interferon (IFN)-γ, together with increased levels of glutamate, reactive oxygen species, and nitric oxide, causing neurotoxicity. These in turn lead to a downregulation of neurotrophic factors, such as vascular endothelial growth factor (VEGF) and glial cell line-derived neurotrophic factor (GDNF) (34), as detailed below. In ALS, activated microglia secrete IL-1α, TNF-α, and complement component 1q (C1q), known to induce neurotoxic reactive astrocytes (35). Abrogation of IL-1α, TNF-α, and C1q was shown to reduce astrogliosis and extend mSOD1 mouse survival. In the absence of reactive astrocytes, MN death is significantly delayed (36). The major mechanisms leading to MN degeneration are summarized in .
Reactive astrocytes promote MN degeneration in ALS. Astrocytes are known to have an aberrant and reactive profile in ALS, releasing several soluble toxic factors and inflammatory mediators that render MNs more susceptible to degeneration. Astrocytes respond (more...)
Aberrant TDP-43 aggregation, a pathological hallmark of both ALS and frontotemporal dementia, was found in astrocytes and shown to contribute to neurodegeneration through cell-specific mechanisms (37). Astrocytes expressing mutated C9ORF72 show a deficient expression of antioxidant proteins, such as SOD1, SOD2 and peroxiredoxins (38). Altogether, astrocytes in ALS not only have a reactive and inflammatory phenotype, but also show impaired protective functions, which may be even exacerbated by the activation of other glial cells such as microglia (35). All these features indicate that astrocytes participate in the control and maintenance of homeostatic balance but, when dysregulated, they lead to neuroinflammation and MN death, thus supporting astrocyte’s key role in the onset and progression of ALS. It is not known if the appearance of an aberrant astrocyte signature previous to ALS symptom onset, in the brain cortex, results from intrinsic cell deficiencies or whether it is determined by MN paracrine pathological signaling. Dysregulated GFAP, S100B, and the marker of proliferation, Ki-67, in immature cortical astrocytes of mSOD1 pups agree with the first hypothesis. Identification of disease-specific astrocytic subpopulations will have a high impact on the understanding of their pathological role in ALS, and on their targeting toward the recovery of a neuroprotective phenotype.
GLUTAMATE AND GABA TRANSPORTERS
Alterations in excitatory neurotransmission appear to play a role in ALS. Hyperexcitability has been observed in sALS and fALS patients before the onset of symptoms, and also in presymptomatic mSOD1 mouse models. However, other studies showed hypoexcitability, rather than hyperexcitability, prior to degeneration (1, 37, 39). Thus, it is still unclear whether hyperexcitability leads to MN degeneration or if it is a compensatory mechanism resulting from MN loss. Astrocytes sustain homeostatic levels of extrasynaptic glutamate within the synaptic cleft to control synaptic transmission, mainly through specific glutamate transporters, such as GLAST and GLT-1 (40). The GLT-1 transporter is found exclusively in astroglia, both in brain and SC, and is responsible for the uptake of nearly 90% of the glutamate. One of the proposed mechanisms for MN death in ALS is glutamate-mediated excitotoxicity (1), since impaired glutamate clearance was shown in astrocytes expressing mSOD1 and TDP-43, suggesting a common pathological feature in ALS () (41, 42).
Glutamate-mediated excitotoxicity in ALS. A. In ALS, the astrocytic glutamate transporter GLT-1 is downregulated, leading to an impaired glutamate uptake, and the TNF-α/TNFR1/NF-κB pathway modulates its expression levels (48). Membralin (more...)
Defects in glutamate uptake by GLT-1 were found in the SC of ALS patients in regions of MN loss (43), in mSOD1 rodents (44), and in TDP-43 mice (45). Several pathways have been implicated in the modulation of GLT-1 levels: (i) TNF-α and downstream NF-κB signaling have been shown to suppress GLT-1 expression (46); (ii) the specific deletion of the ER-component membralin causes a dramatic accumulation of extracellular glutamate, inducing MN glutamatergic toxicity (47); and (iii) decreased GLT-1 expression and glutamate uptake also occur as result of increased astrocyte elevated gene-1 (AEG-1) in mSOD1 astrocytes (48). Moreover, GLT-1 cleavage may derive from the action of caspase-3, with accumulation of a sumoylated GLT-1 C-terminus fragment early on the disease, causing astrocyte phenotypic aberrancies and release of neurotoxic factors (49). Modulation of GLT-1 to potentially prevent excitotoxicity has been attempted. Although, SC focal restoration of GLT-1 expression in astrocytes was not effective (50), enhanced GLT-1 translation by LDN/OSU-0212320 delayed MN function decline and extended the lifespan of mSOD1 mice (51). MC1568, an inhibitor of the enzymes Class-II histone deacetylases, restored GLT-1 expression and glutamate uptake in the SC of mSOD1 mice, but did not prolong their survival (52). Activation of the metabotropic glutamate receptors (mGluRs), mGluR1 and mGluR5, leads to increased intracellular Ca2+ and facilitates glutamate transport (53). In line with this, when the overexpression of dysfunctional mGluR1 and mGluR5 in reactive SC astrocytes from ALS patients and mSOD1 mice was reduced, it prevented excessive glutamate release, improved the function of MNs, astrocytes and microglia, and increased animal survival (54, 55). Deficient glutamate uptake was linked to altered mGluR5-mediated Ca2+ signaling profile (56). By restoring Ca2+ oscillations in astrocytes from mSOD1 rats, mGluR5-mediated glutamate uptake was recovered () (57). While glutamate is the major excitatory neurotransmitter in the CNS, gamma-aminobutyric acid (GABA) and glycine are the main inhibitory neurotransmitters. Raiteri et al. have shown that the activation of a glycine transporter on SC MNs caused enhanced glutamate release in a mouse model of ALS (58). Moreover, in the SC glutamatergic synaptic boutons of mSOD1 mice, the impact of synaptic vesicle exocytosis on the trafficking of nerve terminal GABA transporter-1 (GAT-1) and of type-1/2 glycine (Gly) transporters (GlyT-1/2) was studied by monitoring membrane expression and function of these transporters. It was observed that the enhanced exocytosis in mSOD1 mice boosts heterotransporter membrane expression, which evokes excessive glutamate release () (59). GlyT1/2 and GAT1 are widely expressed in astrocytes and gliosomes (60), together with GABA and glutamate transporters. GABA-induced release of glutamate from SC gliosomes is enhanced in mSOD1 mice (61). Astrocytes were also shown to release higher levels of glutamate, through cystine/glutamate antiporters, in response to oxidative stress (62). SOD1 mutation was shown to reduce intracellular lactate levels and its secretion by astrocytes (63). Studies linking ALS with gut microbiota composition identified that elevated symbiotic Cyanobacteria could promote the elevation of glutamate, in contrast to the general Lactobacillus, Bifidobacterium, and Odoribacter––all known to metabolize glutamate (64). These findings open an all-new window of opportunities to characterize microbiota changes as ALS biomarkers and microbial strategies to improve health status quality of ALS patients.
DYSREGULATED AUTOCRINE/PARACRINE SIGNALING MECHANISMS
Astrocytes have a unique form of excitability, which is characterized by intracellular Ca2+ oscillations or waves in response to physiological and pathophysiological signals. Intracellular Ca2+ elevation is triggered by several mechanisms, such as: (i) activation of Gq-protein-coupled receptors (GPCRs); (ii) GABAB receptor (Gi-coupled GPCRs) activation (65); and (iii) transient receptor potential (TRP) channels (66). Spatially restricted Ca2+ transients in astrocyte microdomains are associated with mitochondria (67). Altogether, they promote the release of gliotransmitters, such as glutamate, D-serine, GABA and neurotoxic factors (68).
The pathogenic potential of Ca2+ dysregulation in astrocytes may account for disease progression. ALS astrocytes show mitochondrial functional deficiencies and impaired Ca2+ homeostasis that promotes MN degeneration (69). In the SC of young mSOD1 mice, the enhanced expression of mGluR5 makes astrocytes vulnerable to glutamate, and causes persistent elevation of intracellular Ca2+ concentrations, which are reverted by Bcl-XL, and protein kinase C epsilon () (57, 70). Administration of the mGluR5 antagonist 2-methyl-6-(phenylethynyl)pyridine (MPEP) slowed astrocyte degeneration, delayed disease onset and extended mSOD1 mouse survival (71). Also, purinergic stimulation of SC and cortical SOD1-expressing astrocytes caused ER Ca2+ accumulation and abnormal Ca2+ signaling (72). The increased expression of Cx43 in mSOD1 mice also has a significant impact on Ca2+ signaling (73). Intracellular Ca2+ increase in astrocytes leads to the release of gliotransmitters, including glutamate, D-serine, GABA, brain derived neurotrophic factor (BDNF), as well as neurotoxic factors (68). Since astrocytes in ALS reveal many pathogenic changes, such as disrupted receptor-mediated Ca2+ signaling and mitochondrial functional deficiencies, it is anticipated that an impaired gliotransmitter release from ALS astrocytes will play a major role in MN pathology.
The storage and release of bioactive molecules by astrocytes involve mechanisms of exocytosis, diffusion through plasma membrane channels, and translocation by plasma membrane transporters. The soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-dependent vesicular exocytotic release is one of the major pathways for astrocyte secretion. ER Ca2+ release induces elevated ATP in mSOD1 astrocytes, which can be inhibited by the overexpression of dominant-negative SNARE to prevent toxicity to MNs and delay disease onset in mSOD1 mice (72). Similarly, pharmacological inhibition of P2X7 receptor abolished astrocyte toxicity towards MNs through degradation of extracellular ATP in mSOD1 mice (74, 75). Since P2X7 receptors form pores under pathophysiological conditions, P2X7 may function as membrane channels that allow the release of glutamate or toxic agents, thus accounting for ALS progression ().
Neurotrophic factors, in particular neurotrophins, are crucial for neuronal differentiation, maturation and survival, as well as for the modulation of synaptic transmission and plasticity (76). They are also potential therapeutic targets for neurodegenerative disorders, such as ALS (77). The neurotrophin family is composed of four members: nerve growth factor (NGF), BDNF, neurotrophin-3 (NT-3) and neurotrophin-4 (NT-4). BDNF is abundantly expressed in the CNS, where it supports neuronal survival (e.g., MNs) (77). SC astrocytes from mSOD1 mice respond to HMGB1 by decreasing BDNF and GDNF production, in contrast to wild-type astrocytes (78). Also, as previously mentioned, astrocytes exposed to the CSF of ALS patients, besides releasing neurotoxic factors, release lower levels of VEGF and GDNF (34).
ALS pathophysiology is intimately related with neuroinflammatory processes, which include the release of both neuroprotective and/or neurotoxic factors that play a role in MN pathology (79). Several studies have shown that TGF-β signaling is involved in ALS and that TGF-β1 release from astrocytes accelerates disease progression in ALS mice (23). Extracellular vesicles with ~40–160 nm, denominated exosomes, are released from mSOD1 astrocytes and were shown to contain mutant SOD1 and dysregulated cargo in miRNAs, accounting for MN pathology and homeostatic imbalance (80, 81). miRNAs are small non-coding RNAs that control posttranscriptional expression of target genes (82). Dying neurons in ALS release miRNAs, such as miRNA(miR)-218, that can change the phenotype of astrocytes into a reactive one and cause the downregulation of GLT-1 (83). In most cases, exosomal cargo in miRNAs recapitulate their cell of origin (84, 85). Dysregulated expression of miRNAs was found in ALS (86, 87) and proposed as biomarkers (88). Upregulation of miR-155 was identified in fALS and sALS patients, as well as in the SC of mSOD1 mice, in pre-symptomatic and symptomatic stages (17). In contrast, a decreased cargo in miR-494-3p was found in C9ORF72 astrocyte-derived exosomes with harmful consequences in neurite network in ALS (89). Depleted levels of miR-146a were also recognized in exosomes from the cortical astrocytes of mSOD1 mice (15), and its cellular replenishment abrogated the astrocyte aberrant phenotype, characterized by increased S100B and Cx43 levels, together with decreased GFAP, while leading to a secretome with paracrine neuroprotective properties (81). Exosomes from both cortical and spinal mSOD1 astrocytes were deficient in miR-155, miR-21 and miR-146a (12). Exosomes with low levels of miRNAs may lead to paracrine dysregulation and dysfunction of recipient cells, while also activating immune-associated cells (90). Exosomes may serve as potential therapeutic targets in ALS and prognostic markers for therapy in precision medicine through patient stratification.
CONCLUSION
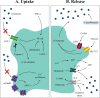
The role of dysfunctional astrocytes in the pathogenesis of ALS indicates that astrocytes may be targeted with strategies for their revival. These strategies may include direct intervention on astrocytes with modulatory medicines, exosomes and miRNA-based therapies, or their replacement (). Considering the first approach, activation of the nuclear factor erythroid 2–related factor (Nrf2) was shown to increase glutathione secretion; although some beneficial effects were observed on glial reactivity, it did not affect survival in mSOD1 mouse models (4). Reduction of reactive oxygen species production has been attempted, but again with no effective benefits (91). Another approach was the overexpression of MHC-I in MNs to enhance their resistance to the toxic factors released by the reactive astrocytes; this approach enhanced the survival of mSOD1 mice (33). As the aberrant astrocytes are associated with inflammatory and immune mechanisms (15, 17), modulation of such mechanisms with specific miRNA-based strategies may prevent cell-to-cell paracrine dysregulation and MN degeneration (81, 86).
Targeting astrocytes for therapy. For functional recovery, astrocyte intervention strategies for ALS may include the modulation of astrocytic activity by using medicines or exosomes, astrocyte-based cell transplantation, and astrosomes (98). The switch (more...)
The use of patient-derived astrocytes by reprogramming techniques brought new possibilities of therapeutic intervention, mainly because drug testing can be done in cells from sALS patients (92, 93). Transplantation of glial restricted precursor cells (94), combined with strategies capable of defending these cells from local toxicity (4), may represent innovative therapeutic approaches for ALS. Transplantation of neural progenitor cells expressing GDNF into the motor cortex of mSOD1 rats showed promise in extending their survival (95). Moreover, Armada-Moreira and colleagues developed an artificial astrocyte (“astrosome”) capable of scavenging hydrogen peroxide and ammonia, by using platinum nanoparticles as artificial enzymes, as well as enzymes capable of glutamate degradation (96–98). Therefore, these microreactors have the potential to provide a therapeutic approach for several neurological diseases, such as ALS, in which oxidative stress and excitotoxicity are observed. We now have the possibility to work with human astrocytes differentiated from sALS and fALS patients and soon it will be possible to identify new targets and stratify patient astrocyte phenotypes, by using 3D microfluidic system models, and test promising therapeutics. Ultimately, this will provide a better understanding of the contribution of astrocytes in ALS, and how we might apply novel therapeutic strategies aimed at producing the revival of astrocytes, or even their replacement, and help in halting, or at least delaying ALS progression.
Acknowledgment: This work was supported by project funding from Fundação para a Ciência e para a Tecnologia (FCT) to SHV (PTDC/BTM-SAL/32147/2017), AMS (PTDC/MED-FAR/30933/2017) and to DB (PTDC/MED-NEU/31395/2017; LISBOA-01-0145-FEDER-031395, and UID/DTP/04138/2018), as well as from Santa Casa da Misericórdia (ALS Research Grant ELA-2015-002 to DB). This project has received funding from H2020-WIDESPREAD-05-2017-Twinning (EpiEpinet) under grant agreement No. 952455. SP (SFRH/BD/147277/2019) is a FCT PhD grant recipient.
Conflict of interest: The authors declare no potential conflict of interest with respect to research, authorship and/or publication of this manuscript.
Copyright and permission statement: The authors confirm that the materials included in this chapter do not violate copyright laws. Where relevant, appropriate permissions have been obtained from the original copyright holder(s), and all original sources have been appropriately acknowledged or referenced.
REFERENCES
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
Gomes C, Cunha C, Nascimento F, Ribeiro JA, Vaz AR, Brites D. Cortical Neurotoxic Astrocytes with Early ALS Pathology and miR-146a Deficit Replicate Gliosis Markers of Symptomatic SOD1G93A Mouse Model.
Mol Neurobiol. 2019;56(3):2137–58.
https://doi.org/10.1007/s12035-018-1220-8
. [
PubMed: 29995256]
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
Fujita K, Kato T, Yamauchi M, Ando M, Honda M, Nagata Y. Increases in fragmented glial fibrillary acidic protein levels in the spinal cords of patients with amyotrophic lateral sclerosis.
Neurochem Res. 1998;23(2):169–74.
https://doi.org/10.1023/A:1022476724381
. [
PubMed: 9475511]
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
Kunze A, Lengacher S, Dirren E, Aebischer P, Magistretti PJ, Renaud P. Astrocyte-neuron co-culture on microchips based on the model of SOD mutation to mimic ALS.
Integr Biol (United Kingdom). 2013;5(7):964–75.
https://doi.org/10.1039/c3ib40022k
. [
PubMed: 23695230]
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
- 36.
- 37.
- 38.
- 39.
- 40.
- 41.
Wallis N, Lau CL, Farg MA, Atkin JD, Beart PM, O’Shea RD. SOD1 Mutations Causing Familial Amyotrophic Lateral Sclerosis Induce Toxicity in Astrocytes: Evidence for Bystander Effects in a Continuum of Astrogliosis.
Neurochem Res. 2018;43(1):157–70.
https://doi.org/10.1007/s11064-017-2385-7
. [
PubMed: 28861673]
- 42.
Barton SK, Lau CL, Chiam MDF, Tomas D, Muyderman H, Beart PM, et al. Mutant TDP-43 Expression Triggers TDP-43 Pathology and Cell Autonomous Effects on Primary Astrocytes: Implications for Non-cell Autonomous Pathology in ALS.
Neurochem Res. 2020;45(6):1451–9.
https://doi.org/10.1007/s11064-020-03048-5
. [
PubMed: 32410044]
- 43.
- 44.
- 45.
- 46.
- 47.
- 48.
Yin X, Wang S, Qi Y, Wang X, Jiang H, Wang T, et al. Astrocyte elevated gene-1 is a novel regulator of astrogliosis and excitatory amino acid transporter-2 via interplaying with nuclear factor-κB signaling in astrocytes from amyotrophic lateral sclerosis mouse model with hSOD1G93A mutation.
Mol Cell Neurosci. 2018;90:1–11.
https://doi.org/10.1016/j.mcn.2018.05.004
. [
PubMed: 29777762]
- 49.
- 50.
- 51.
- 52.
- 53.
- 54.
Milanese M, Giribaldi F, Melone M, Bonifacino T, Musante I, Carminati E, et al. Knocking down metabotropic glutamate receptor 1 improves survival and disease progression in the SOD1G93A mouse model of amyotrophic lateral sclerosis.
Neurobiol Dis. 2014;64:48–59.
https://doi.org/10.1016/j.nbd.2013.11.006
. [
PubMed: 24361555]
- 55.
- 56.
Vermeiren C, De Hemptinne I, Vanhoutte N, Tilleux S, Maloteaux JM, Hermans E. Loss of metabotropic glutamate receptor-mediated regulation of glutamate transport in chemically activated astrocytes in a rat model of amyotrophic lateral sclerosis.
J Neurochem. 2006;96(3):719–31.
https://doi.org/10.1111/j.1471-4159.2005.03577.x
. [
PubMed: 16371010]
- 57.
Vergouts M, Doyen PJ, Peeters M, Opsomer R, Hermans E. Constitutive downregulation protein kinase C epsilon in hSOD1G93A astrocytes influences mGluR5 signaling and the regulation of glutamate uptake.
Glia. 2018;66(4):749–61.
https://doi.org/10.1002/glia.23279
. [
PubMed: 29266405]
- 58.
- 59.
Milanese M, Bonifacino T, Fedele E, Rebosio C, Cattaneo L, Benfenati F, et al. Exocytosis regulates trafficking of GABA and glycine heterotransporters in spinal cord glutamatergic synapses: A mechanism for the excessive heterotransporter-induced release of glutamate in experimental amyotrophic lateral sclerosis.
Neurobiol Dis. 2015;74:314–24.
https://doi.org/10.1016/j.nbd.2014.12.004
. [
PubMed: 25497732]
- 60.
Raiteri L, Stigliani S, Usai C, Diaspro A, Paluzzi S, Milanese M, et al. Functional expression of release-regulating glycine transporters GLYT1 on GABAergic neurons and GLYT2 on astrocytes in mouse spinal cord.
Neurochem Int. 2008;52(1):103–12.
https://doi.org/10.1016/j.neuint.2007.04.027
. [
PubMed: 17597258]
- 61.
Milanese M, Zappettini S, Jacchetti E, Bonifacino T, Cervetto C, Usai C, et al. In vitro activation of GAT1 transporters expressed in spinal cord gliosomes stimulates glutamate release that is abnormally elevated in the SOD1/G93A(+) mouse model of amyotrophic lateral sclerosis.
J Neurochem. 2010;113(2):489–501.
https://doi.org/10.1111/j.1471-4159.2010.06628.x
. [
PubMed: 20132478]
- 62.
Kazama M, Kato Y, Kakita A, Noguchi N, Urano Y, Masui K, et al. Astrocytes release glutamate via cystine/glutamate antiporter upregulated in response to increased oxidative stress related to sporadic amyotrophic lateral sclerosis.
Neuropathology. 2020;40(6):587–98.
https://doi.org/10.1111/neup.12716
. [
PubMed: 33305472]
- 63.
Madji Hounoum B, Mavel S, Coque E, Patin F, Vourc’h P, Marouillat S, et al. Wildtype motoneurons, ALS-Linked SOD1 mutation and glutamate profoundly modify astrocyte metabolism and lactate shuttling.
Glia. 2017;65(4):592–605.
https://doi.org/10.1002/glia.23114
. [
PubMed: 28139855]
- 64.
- 65.
- 66.
- 67.
- 68.
- 69.
- 70.
Martorana F, Brambilla L, Valori CF, Bergamaschi C, Roncoroni C, Aronica E, et al. The BH4 domain of Bcl-X L rescues astrocyte degeneration in amyotrophic lateral sclerosis by modulating intracellular calcium signals.
Hum Mol Genet. 2012;21(4):826–40.
https://doi.org/10.1093/hmg/ddr513
. [
PubMed: 22072391]
- 71.
- 72.
- 73.
- 74.
- 75.
- 76.
Sebastião AM, Assaife-Lopes N, Diógenes MJ, Vaz SH, Ribeiro JA. Modulation of brain-derived neurotrophic factor (BDNF) actions in the nervous system by adenosine A2A receptors and the role of lipid rafts.
Biochimica et Biophysica Acta (BBA) - Biomembranes. 2011;1808(5):1340–1349.
https://doi.org/10.1016/j.bbamem.2010.06.028
. [
PubMed: 20603099]
- 77.
- 78.
- 79.
- 80.
Basso M, Pozzi S, Tortarolo M, Fiordaliso F, Bisighini C, Pasetto L, et al. Mutant Copper-Zinc Superoxide Dismutase (SOD1) Induces Protein Secretion Pathway Alterations and Exosome Release in Astrocytes: implications for disease spreading and motor neuron pathology in Amyotrophic Lateral Sclerosis.
J Biol Chem. 2013;288(22):15699–711.
https://doi.org/10.1074/jbc.M112.425066
. [
PMC free article: PMC3668729] [
PubMed: 23592792]
- 81.
- 82.
- 83.
- 84.
- 85.
- 86.
- 87.
Freischmidt A, Müller K, Zondler L, Weydt P, Volk AE, Božič AL, et al. Serum microRNAs in patients with genetic amyotrophic lateral sclerosis and pre-manifest mutation carriers.
Brain. 2014;137(11):2938–50.
https://doi.org/10.1093/brain/awu249
. [
PubMed: 25193138]
- 88.
- 89.
- 90.
- 91.
- 92.
- 93.
- 94.
- 95.
Thomsen GM, Avalos P, Ma AA, Alkaslasi M, Cho N, Wyss L, et al. Transplantation of Neural Progenitor Cells Expressing Glial Cell Line-Derived Neurotrophic Factor into the Motor Cortex as a Strategy to Treat Amyotrophic Lateral Sclerosis.
Stem Cells. 2018;36(7):1122–31.
https://doi.org/10.1002/stem.2825
. [
PubMed: 29656478]
- 96.
Armada-Moreira A, Taipaleenmäki E, Baekgaard-Laursen M, Schattling PS, Sebastião AM, Vaz SH, et al. Platinum Nanoparticle-Based Microreactors as Support for Neuroblastoma Cells.
ACS Appl Mater Interfaces. 2018;10(9):7581–92.
https://doi.org/10.1021/acsami.7b10724
. [
PubMed: 29083859]
- 97.
- 98.



 1,2
1,2