Summary
Clinical characteristics.
Salih myopathy is characterized by muscle weakness (manifest during the neonatal period or in early infancy) and delayed motor development; children acquire independent walking between ages 20 months and four years. In the first decade of life, global motor performance is stable or tends to improve. Moderate joint and neck contractures and spinal rigidity may manifest in the first decade but become more obvious in the second decade. Scoliosis develops after age 11 years. Cardiac dysfunction manifests between ages five and 16 years, progresses rapidly, and leads to death between ages eight and 20 years, usually from heart rhythm disturbances.
Diagnosis/testing.
The diagnosis is established in a proband by identification of biallelic pathogenic variants in the first three M-line-encoding exons (Mex1, Mex2, and Mex3) of TTN, the only gene in which pathogenic variants are known to cause Salih myopathy.
Management.
Treatment of manifestations: Care, best provided by a multidisciplinary team, includes stretching exercises and physical therapy; assistive mechanical devices for sitting and ambulation as needed; and appropriate technical support in educational settings. Treat heart failure and cardiac arrhythmia as soon as they are evident. Cardiac transplantation may be considered for progressive dilated cardiomyopathy and heart failure refractory to medical therapy.
Prevention of secondary complications: Annual influenza vaccine and other respiratory infection-related immunizations are advised. Aggressive treatment of lower-respiratory tract infections.
Surveillance: Electrocardiogram (EKG), 24-hour Holter EKG, and echocardiogram every six months beginning at age five years. Annual evaluation of respiratory function beginning at age 10 years. Clinical examination and x-ray as needed for orthopedic complications (e.g., foot deformity, joint contractures, spinal deformity).
Agents/circumstances to avoid: Ibuprofen in those with congestive heart failure.
Genetic counseling.
Salih myopathy is inherited in an autosomal recessive manner. The parents of an affected child are obligate heterozygotes (i.e., carriers of one pathogenic variant) and are asymptomatic. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk relatives and prenatal testing for a pregnancy at increased risk are possible if the pathogenic variants in the family have been identified.
Diagnosis
Suggestive Findings
Salih myopathy should be suspected in individuals with the following clinical, laboratory, electrophysiologic, imaging, and histopathology findings.
Clinical
- Muscle weakness manifesting during the neonatal period or in early infancy
- Delayed motor milestones but normal cognitive development
- Muscle weakness of limb-girdle distribution, myopathic face, variable degree of ptosis, and relative calf muscle hypertrophy
- Development of dilated cardiomyopathy between ages five and 16 years
- Major heart rhythm disturbances leading to sudden death before age 20 years
Laboratory. Serum creatine kinase (CK) is marginally to moderately increased (1.5-7x normal).
Electrophysiologic
- Electrocardiography. Left axis deviation (left anterior fascicular block) can be seen as early as age four years (Figure 1). With the onset of dilated cardiomyopathy, rhythm disturbances can include polymorphic premature ventricular complexes, bigeminism and trigeminism, couplets, triplets, atrioventricular heart block, atrioventricular nodal reentrant tachycardia, premature atrial complexes, premature ventricular complexes, and ventricular tachycardia.
- Electromyography shows myopathic features (low-amplitude polyphasic potentials of short duration).
- Nerve conduction studies are normal.
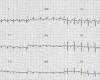
Figure 1.
Electrocardiogram at age four years showing left axis deviation (left anterior fascicular block)
Echocardiogram reveals, at the onset of cardiomyopathy, reduced function of the left ventricle and dilatation and global hypokinesia without wall hypertrophy. Later, dilatation involves the left atrium and ventricle, subsequently affecting all chambers.
Skeletal muscle biopsy. The following findings help distinguish Salih myopathy from congenital muscular dystrophy (CMD) and other congenital myopathies. Muscle biopsy sections should be examined for histology, histochemistry, immunohistochemistry, and electron microscopy:
- Histology of skeletal muscle reveals a pattern compatible with congenital myopathy (Figure 2): mild variation in fiber size, abundant centrally located nuclei, no increase in connective tissue before age six years, and mild endomysial fibrosis after age six years.Oxidative stains reveal multiple small lesions of reduced or absent oxidative activity with poorly defined boundaries.Myofibrillar ATpase staining shows remarkable type 1 fiber predominance (>90%). In one individual, massive muscle fiber loss was seen in a second biopsy taken at age ten years [Carmignac et al 2007].
- Immunohistochemistry of skeletal muscle shows normal expression of dystrophin, laminin α2 chain (merosin), integrin α7, α- and β-dystroglycan, desmin, emerin, and the sarcoglycans α (adhalin), β, γ, and δ.
- Electron microscopy of skeletal muscle (Figure 3) highlights the "minicore-like" lesions seen on histology and reveals multiple foci of sarcomere disruption and mitochondria depletion.


Establishing the Diagnosis
The diagnosis of Salih myopathy is established in a proband by identification of biallelic pathogenic variants in M-line-encoding exons Mex1, Mex2, and Mex3* of TTN on molecular genetic testing (see Table 1).
*Note: The last six exons of TTN (359 to 364 in the LRG [NG_011618.3] numbering that numbers the exons sequentially along the chromosome; C-terminal domain) encode the part of titin that spans the sarcomere M-line; these exons are called Mex1-Mex6 for "M-line-encoding exons 1 through 6." For mapping of these exons to other transcripts, see Molecular Genetics.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of Salih myopathy is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with myopathy and/or cardiomyopathy are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of Salih myopathy, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of TTN detects small intragenic deletions/insertions and missense, nonsense, and splice site variants; typically, exon or whole-gene deletions/duplications are not detected. Perform sequence analysis of select exons (Mex1, Mex2, Mex3) first. If only one or no pathogenic variant is found, perform TTN sequence analysis of the remaining exons and deletion/duplication analysis.Note: If biallelic TTN pathogenic variants are not identified with the above testing, RNA sequencing to identify variants that alter splicing and/or expression and western blot analysis can be considered; however, such testing may not be available on a clinical basis [Savarese et al 2018a].
- A multigene panel that includes TTN and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. Of note, given the rarity of Salih myopathy, some panels for myopathy and/or cardiomyopathy may not include this gene. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by myopathy, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Salih Myopathy
Clinical Characteristics
Clinical Description
Salih myopathy is characterized by muscle weakness manifest during the neonatal period or in early infancy, and delayed motor development. Children acquire independent walking between ages 20 months and four years. In the first decade of life, global motor performance is stable or tends to improve. During this period skeletal muscle involvement mainly manifests as difficulty in running, climbing stairs, and rising from a sitting position. Those who survive childhood remain ambulant, with or without support, and maintain normal cognitive function.
Musculoskeletal. Moderate joint and neck contractures and spinal rigidity may appear in the first decade but become more obvious in the second decade. Respiratory function tests show a moderate restrictive pattern. Scoliosis develops after age 11 years.
Cardiac dysfunction manifests between ages five and 16 years and progresses rapidly, leading to death between ages eight and 20 years. Heart rhythm disturbances are the major cause of sudden death and their frequency and severity suggest primary involvement of the conduction system.
Electrocardiography (EKG) is very helpful in signaling the occurrence of cardiac involvement. Left axis deviation (left anterior fascicular block) can be seen as early as age four years (Figure 1). With the onset of dilated cardiomyopathy (between ages 5 and 16 years), major rhythm disturbances become evident on EKG and Holter monitoring, including polymorphic premature ventricular complexes, bigeminism and trigeminism, couplets, triplets, atrioventricular heart block, atrioventricular nodal reentrant tachycardia, premature atrial complexes, premature ventricular complexes, and ventricular tachycardia.
Echocardiogram reveals, at the onset of cardiomyopathy, reduced function of the left ventricle and dilatation and global hypokinesia without wall hypertrophy. Later, dilatation involves the left atrium and ventricle, subsequently affecting all chambers and leading to congestive heart failure.
Radionuclide angiography using MUGA (multi-gated acquisition) scan reveals the deteriorating ventricular function with reduction of the left ventricular ejection fraction followed by reduction of the right ventricle ejection fraction.
Heart muscle biopsies (taken from 2 individuals) showed increased interstitial fibrosis compatible with dilated cardiomyopathy [Carmignac et al 2007]. Oxidative staining was normal without focal oxidative defects or significant disarray of the cardiomyocyte structure, in contrast to the classic observation in hypertrophic cardiomyopathy.
Heterozygotes. In contrast to individuals with heterozygous pathogenic variants in TTN associated with Udd distal myopathy, the heterozygous parents of individuals with Salih myopathy remain asymptomatic with no cardiac or muscle disorder (Figure 4).
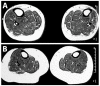
Figure 4.
Mid-calf muscle MRI of parents of a proband at age (A) 55 years and (B) 44 years were normal and showed no fatty degeneration of the anterior tibial muscles.
Genotype-Phenotype Correlations
No genotype-phenotype correlations are known.
Nomenclature
Salih myopathy was initially referred to as Salih congenital muscular dystrophy [Salih et al 1998, Subahi 2001]. Subsequently, it was renamed Salih myopathy [Fukuzawa et al 2008, Pernigo et al 2010].
Prevalence
Salih myopathy is thought to be rare. It has been described in consanguineous families of Arab descent originating from Sudan and Morocco. The actual prevalence is unknown.
Genetically Related (Allelic) Disorders
Limb-girdle muscular dystrophy type 2J (LGMD2J), which is allelic to Salih myopathy and is caused by mutation of the C terminus of TTN, has a later onset (childhood or later) than Salih myopathy.
Other phenotypes known to be associated with TTN include the following autosomal dominant disorders:
- Dilated cardiomyopathy (DCM)
For comprehensive reviews of TTN-related disorders, see Savarese et al [2016] and Hackman et al [2017].
Differential Diagnosis
Table 2.
Early-Onset Muscle Disorders to Consider in the Differential Diagnosis of Salih Myopathy
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Salih myopathy, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis of Salih Myopathy
Treatment of Manifestations
Treatment involves prompt management of disease manifestations using a multidisciplinary approach that includes specialists in pediatric neurology, physiotherapy, occupational therapy, orthopedics, cardiology, and pulmonology.
Stretching exercises and physical therapy help prevent contractures and promote mobility. Assistive mechanical devices including orthotics, canes, and walkers can be used as needed.
Individuals with Salih myopathy have normal cognition: technical support should be provided in the school environment. Stimulation and emotional support can improve school performance and sense of social involvement.
Parents and/or caregivers should be made aware of the symptoms of heart failure, arrhythmia (including presyncope and syncope), and thromboembolic disease, and of the need to urgently seek medical care when any of these symptoms appear.
Training of caregivers in cardiopulmonary resuscitation may be suggested once the symptoms of cardiomyopathy start.
Adequate posture should be maintained when lying prone and sitting. Garchois brace (made of plexidur, a rigid but light and heat-deformable material) is used to reduce the degree of deformity and slow the progression of scoliosis [Wang et al 2010].
Cardiac transplantation should be considered for progressive dilated cardiomyopathy and heart failure refractory to medical therapy.
Prevention of Secondary Complications
Annual influenza vaccine and other respiratory infection-related immunizations are advised.
Lower respiratory tract infections should be treated aggressively when they occur.
Surveillance
Table 4.
Recommended Surveillance for Individuals with Salih Myopathy
Agents/Circumstances to Avoid
Ibuprofen (Brufen®):
- Give with care in those with evidence of cardiomyopathy. A patient who had reduced left ventricular ejection fraction developed edema following its administration [Subahi & Salih, unpublished observation].
- Avoid in those with congestive heart failure.
Evaluation of Relatives at Risk
Early diagnosis of at-risk sibs by clinical examination and/or molecular genetic testing is important in order to monitor motor development and cardiac function so that treatment can be instituted promptly.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
If a fetus is diagnosed prenatally with Salih myopathy, special considerations are needed at and following delivery since muscle weakness may manifest during the neonatal period.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Salih myopathy is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., carriers) of one Salih myopathy-related TTN pathogenic variant.
- Heterozygotes (carriers) are asymptomatic. Note: In contrast to individuals with heterozygous pathogenic variants in TTN associated with Udd distal myopathy, the parents of individuals with Salih myopathy remain asymptomatic with no cardiac or muscle disorder (Figure 4).
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic.
Offspring of a proband. Individuals with Salih myopathy are not known to have reproduced.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a TTN pathogenic variant.
Carrier Detection
Carrier testing for at-risk family members requires prior identification of the TTN pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are carriers or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the TTN pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for Salih myopathy are possible.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Muscular Dystrophy Association (MDA) - USAPhone: 833-275-6321
- Muscular Dystrophy CanadaCanadaPhone: 800-567-2873Email: info@muscle.ca
- Muscular Dystrophy UKUnited KingdomPhone: 0800 652 6352
- Congenital Muscle Disease International Registry (CMDIR)The CMDIR is a global partnership of patient advocacy organizations, researchers, and clinicians, all working toward the same goal: to find treatments for congenital muscle disease.CMDIR/Cure CMD
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Salih Myopathy: Genes and Databases
Table B.
OMIM Entries for Salih Myopathy (View All in OMIM)
Molecular Pathogenesis
To date, biallelic pathogenic variants reported to cause Salih myopathy are small frameshift deletions in TTN exons Mex1 and Mex3 in consanguineous families of Moroccan and Sudanese origin [Carmignac et al 2007]. Carriers of these frameshift variants (i.e., heterozygotes) are asymptomatic, presumably as a result of nonsense-mediated decay (NMD) of the mutated mRNA. Since homozygotes survive, the NMD cannot be complete. Some titin protein that lacks the last C-terminal domains is produced. Whether the total decrease of titin protein or the loss of C terminus is more important for the phenotype is not known.
Gene structure. The longest TTN transcript, NM_001267550.1, has 363 exons with a coding capacity of 113,414 bp. TTN has a large number of alternative splicing variants, which can result in confusion in exon and nucleotide numbering in the literature [Bang et al 2001, Guo et al 2010, Savarese et al 2018b]. Transcript variant (N2A) NM_133378.4 is a long transcript with 312 exons that encodes the isoform N2A, the predominant titin isoform in skeletal muscle. For a detailed summary of gene and protein information, see Table A, Gene.
The Mex1 – Mex6 domain exons correspond to the following exons:
- LRG_391 (NG_011618.3): exons 359-364
- NM_133378.4: exons 307-312
- NM_001267550.1: exons 358-363
- NM_001256850.1: exons 307-312
Pathogenic variants. To date, the biallelic variants reported to cause Salih myopathy are small frameshift deletions in TTN exons Mex1 and Mex3 in two consanguineous families of Moroccan and Sudanese origin [Carmignac et al 2007] (Table 2).
Table 5.
Selected TTN Pathogenic Variants
Normal gene product. Titin is the biggest single polypeptide in humans, found in numerous isoforms of varying size. The entire coding region predicts a protein of 38,138 amino acids (4,200 kd). Titin is expressed as several different isoforms, caused by alternative splicing, in different skeletal muscles and in cardiac muscle [Bang et al 2001, Guo et al 2010].
Titin functions as a template in sarcomere assembly and for maintenance of sarcomere integrity. The titin protein is the third myofilament in the sarcomere along with myosin and actin filaments. Titin spans more than one half the length of a sarcomere in heart and skeletal muscle. Structurally different parts of the protein perform distinct functions (mechanical, developmental, and regulatory) [Carmignac et al 2007]. Titin binds and interacts with a large number of other sarcomeric proteins.
Abnormal gene product. The predicted truncated titin proteins resulting from the frameshift variants in Mex1, Mex2, and Mex3 are incorporated into ultrastructurally normal sarcomeres in homozygous affected individuals [Carmignac et al 2007]. Therefore, absence of the last five (Mex2-Mex6) exons is compatible with life but causes this severe congenital disorder. Asymptomatic heterozygous carriers of these titin deletions presumably have sufficient full-length titin to result in normal sarcomere function.
Chapter Notes
Acknowledgments
The authors would like to thank Loida M Sese for secretarial work, and Sayed Taha and Vir Salvador for medical illustration.
Author History
Virginie Carmignac, PhD (2012-present)
Peter Hackman, PhD (2012-present)
Mustafa Salih, MB BS, MPCH, MD, Dr Med Sci, FRCPCH, FAAN (2012-present)
Marco Savarese, PhD (2019-present)
Tiina Suominen, MSc; University of Helsinki (2012-2019)
Bjarne Udd, MD, PhD (2012-present)
Revision History
- 11 April 2019 (sw) Comprehensive update posted live
- 12 January 2012 (me) Review posted live
- 28 October 2011 (mas) Original submission
References
Literature Cited
- Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, Labeit S. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res. 2001;89:1065–72. [PubMed: 11717165]
- Carmignac V, Salih MAM, Quijano-Roy S, Marchand S, Marchand S, Al Rayess MM, Mukhtar MM, Ja U, Labeit S, Guicheney P, Leturcq F, Gautel M, Fardau M, Campbell KP, Richard I, Estournet B, Ferreiro A. C-terminal titin deletions cause a novel early-onset myopathy with fatal cardiomyopathy. Ann Neurol. 2007;61:340–51. [PubMed: 17444505]
- Fukuzawa A, Lange S, Holt M, Vichola A, Carmignac V, Ferreiro A, Udd B, Gautel M. Interchains with titin and myomesin target obscuring and obscuring-like 1 to the M-band-implication for hereditary myopathies. J Cell Sci. 2008;121:1841–51. [PubMed: 18477606]
- Guo W, Bharmal SJ, Esbona K, Greaser ML. Titin diversity--alternative splicing gone wild. J Biomed Biotechnol. 2010;2010:753675. [PMC free article: PMC2843904] [PubMed: 20339475]
- Hackman P, Udd B, Bönnemann CG, Ferreiro A, et al. 219th ENMC International Workshop Titinopathies International database of titin mutations and phenotypes, Heemskerk, The Netherlands, 29 April-1 May 2016. Neuromuscul Disord. 2017;27:396–407. [PubMed: 28214268]
- Nigro V, Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol. 2014;33:1–12. [PMC free article: PMC4021627] [PubMed: 24843229]
- Pernigo S, Fukuzawa A, Bertz M, Holt M, Reif M, Steiner RA, Gautel M. Structural insight into M-band assembly and mechanics from the titin-obscurin-like-1 complex. Proc Natl Acad Sci USA. 2010;107:2908–13. [PMC free article: PMC2814874] [PubMed: 20133654]
- Salih MA, Al Rayess M, Cutshall S, Urtizberca JA, Al-Turaiki MH, Ozo CO, Straub V, Akbar M, Abid M, Andeejani A, Campell KP. A novel form of familial congenital muscular dystrophy in two adolescents. Neuropediatrics. 1998;29:289–93. [PubMed: 10029346]
- Savarese M, Jonson PH, Huovinen S, Paulin L, Auvinen P, Udd B, Hackman P. The complexity of titin splicing pattern in human adult skeletal muscles. Skelet Muscle. 2018a;8:11. [PMC free article: PMC5874998] [PubMed: 29598826]
- Savarese M, Maggi L, Vihola A, Jonson PH, Tasca G, Ruggiero L, Bello L, Magri F, Giugliano T, Torella A, Evilä A, Di Fruscio G, Vanakker O, Gibertini S, Vercelli L, Ruggieri A, Antozzi C, Luque H, Janssens S, Pasanisi MB, Fiorillo C, Raimondi M, Ergoli M, Politano L, Bruno C, Rubegni A, Pane M, Santorelli FM, Minetti C, Angelini C, De Bleecker J, Moggio M, Mongini T, Comi GP, Santoro L, Mercuri E, Pegoraro E, Mora M, Hackman P, Udd B, Nigro V. Interpreting genetic variants in titin in patients with muscle disorders. JAMA Neurol. 2018b;75:557–65. [PMC free article: PMC5885217] [PubMed: 29435569]
- Savarese M, Sarparanta J, Vihola A, Udd B, Hackman P. Increasing role of titin mutations in neuromuscular disorders. J Neuromuscul Dis. 2016;3:293–308. [PMC free article: PMC5123623] [PubMed: 27854229]
- Subahi SA. Distinguishing cardiac features of a novel form of congenital muscular dystrophy (Salih cmd). Pediatr Cardiol. 2001;22:297–301. [PubMed: 11455396]
- Wang CH, Bonnemann CG, Rutkowski A, Sejersen T, Bellini J, Battista V, Florence JM, Schara U, Schuler PM, Wahbi K, Aloysius A, Bash RO, Béroud C, Bertini E, Bushby K, Cohn RD, Connolly AM, Deconinck N, Desguerre I, Eagle M, Estournet-Mathiaud B, Ferreiro A, Fujak A, Goemans N, Iannaccone ST, Jouinot P, Main M, Melacini P, Mueller-Felber W, Muntoni F, Nelson LL, Rahbek J, Quijano-Roy S, Sewry C, Storhaug K, Simonds A, Tseng B, Vajsar J, Vianello A, Zeller R. Consensus statement on standard of care for congenital muscular dystrophies. J Child Neurol. 2010;25:1559–81. [PMC free article: PMC5207780] [PubMed: 21078917]
Publication Details
Author Information and Affiliations
Haartman Institute
University of Helsinki
Helsinki, Finland
Haartman Institute
University of Helsinki
Helsinki, Finland
IFR Santé-STIC
Université de Bourgogne
Dijon, France
The Folkhälsan Institute of Genetics and the Department of Medical Genetics
Haartman Institute
University of Helsinki
Helsinki, Finland
College of Medicine
King Saud University
Riyadh, Saudi Arabia
Publication History
Initial Posting: January 12, 2012; Last Update: April 11, 2019.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Hackman P, Savarese M, Carmignac V, et al. Salih Myopathy. 2012 Jan 12 [Updated 2019 Apr 11]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.



