Summary
Clinical characteristics.
SLC25A24 Fontaine progeroid syndrome is a multisystem connective tissue disorder characterized by poor growth, abnormal skeletal features, and distinctive craniofacial features with sagging, thin skin, and decreased subcutaneous fat suggesting an aged appearance that is most pronounced in infancy and improves with time. Characteristic radiographic features include turribrachycephaly with widely open anterior fontanelle, craniosynostosis, and anomalies of the terminal phalanges. Cardiovascular, genitourinary, ocular, and gastrointestinal abnormalities may also occur. To date, 13 individuals with a molecularly confirmed diagnosis of SLC25A24 Fontaine progeroid syndrome have been described.
Diagnosis/testing.
The diagnosis of SLC25A24 Fontaine progeroid syndrome is established in a proband with suggestive findings and a heterozygous pathogenic variant in SLC25A24 identified by molecular genetic testing.
Management.
Treatment of manifestations: Management, which is largely symptomatic, may be performed by specialists in multiple disciplines, including a craniofacial clinic (involving plastic surgery, neurosurgery, and otolaryngology), cardiology, pulmonology, gastroenterology, and clinical genetics. Some students may benefit from an individualized education plan through their school.
Surveillance: Routine evaluation to assess development of new manifestations and response to ongoing management.
Agents/circumstances to avoid: Contact sports and isometric exercise may need to be restricted if cranial anomalies and/or aortic dilatation are present.
Genetic counseling.
SLC25A24 Fontaine progeroid syndrome is an autosomal dominant disorder typically caused by a de novo pathogenic variant. Risk to future pregnancies is presumed to be low as the proband most likely has a de novo SLC25A24 pathogenic variant. However, given a recurrence risk (~1%) to sibs based on the theoretic possibility of parental germline mosaicism, prenatal and preimplantation genetic testing may be considered.
Diagnosis
No consensus clinical diagnostic criteria for SLC25A24 Fontaine progeroid syndrome have been published.
Suggestive Findings
SLC25A24 Fontaine progeroid syndrome should be suspected in individuals with the following clinical and radiographic findings.
Clinical findings
- Pre- and postnatal growth failure
- Sagging, thin, and translucent skin with decreased subcutaneous fat, contributing to a progeroid appearance, most pronounced in infancy
- Distinctive craniofacial features (Figure 1). Core features recognizable from infancy include turribrachycephaly, short and downslanted palpebral fissures, depressed nasal root, midfacial retrusion, and small and low-set ears.
- Cranial underossification with large anterior fontanelle (Figure 2), frequently presenting with craniosynostosis
- Sparse scalp hair in infancy with low anterior and posterior hairlines transitioning to coarse, unruly hair with multiple hair whorls (see Figure 1)
- Hypertrichosis of face, back, and extensor surfaces
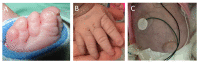
- Umbilical hernia and underdeveloped abdominal wall musculature (Figure 3)
- Digital anomalies including short distal phalanges, cutaneous syndactyly of fingers or toes, and abnormal nail development (see Figure 3)
- Hypoplastic labia majora in females or cryptorchidism in males
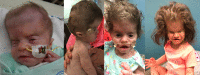
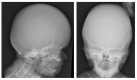
Figure 2.
Skull radiographs from the same child at age two weeks. Note nearly absent calvarial ossification.
Radiographic findings
- Poorly ossified calvarium (see Figure 2), often with craniosynostosis evident on CT
- Short distal phalanges, with or without cutaneous syndactyly
Establishing the Diagnosis
The diagnosis of SLC25A24 Fontaine progeroid syndrome is established in a proband with suggestive findings and a heterozygous pathogenic (or likely pathogenic) variant in SLC25A24 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of a heterozygous SLC25A24 variant of uncertain significance does not establish or rule out the diagnosis of this disorder.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing).
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of SLC25A24 Fontaine progeroid syndrome has not been considered may be more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and imaging findings suggest the diagnosis of SLC25A24 Fontaine progeroid syndrome, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of SLC25A24 is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Typically, if no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications; however, to date such variants have not been identified as a cause of this disorder.
- A craniosynostosis or skeletal disorder multigene panel that includes SLC25A24 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the diagnosis of SLC25A24 Fontaine progeroid syndrome has not been considered because an individual has atypical phenotypic features, comprehensive genomic testing, which does not require the clinician to determine which gene is likely involved, is an option. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in SLC25A24 Fontaine Progeroid Syndrome
Clinical Characteristics
Clinical Description
SLC25A24 Fontaine progeroid syndrome is a multisystem connective tissue disorder characterized by poor growth, abnormal skeletal features, and distinctive craniofacial features with sagging, thin skin and decreased subcutaneous fat suggesting an aged appearance that is most pronounced in infancy and improves with time. Characteristic radiographic features include turribrachycephaly with widely open anterior fontanelle, craniosynostosis, and anomalies of the terminal phalanges. Cardiovascular, genitourinary, ocular, and gastrointestinal abnormalities may also occur.
Prior to identification of the molecular basis of this disorder, clinical findings were published by Gorlin et al [1960], Fontaine et al [1977], and Petty et al [1990]. Subsequent published case reports noted significant clinical overlap particularly with "Petty syndrome" and "Fontaine-Farriaux syndrome" [Castori et al 2009, Braddock et al 2010, Writzl et al 2017]. To date, 13 individuals have been described with a molecularly confirmed diagnosis of SLC25A24 Fontaine progeroid syndrome [Ehmke et al 2017; Writzl et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Legué et al 2020; Author, unpublished data]. The clinical findings discussed in this section are based on these reports (see Table 2).
Note: (1) Of the four individuals with an SLC25A24 pathogenic variant included in the report by Writzl et al [2017], three had previously been reported: Patient 2 in Faivre et al [1999], Patient 3 in Rodríguez et al [1999], and Patient 4 in Castori et al [2009]. (2) Of the five females with an SLC25A24 pathogenic variant and clinical findings suggestive of Gorlin-Chaudhry-Moss syndrome included in the report by Ehmke et al [2017], one (Patient 4) had previously been reported by Adolphs et al [2011].
Table 2.
Select Features of SLC25A24 Fontaine Progeroid Syndrome
Growth
Prenatal growth. All but one affected individual presented with intrauterine growth restriction [Ehmke et al 2017]. Oligohydramnios is also commonly noted in the third trimester.
Z scores for birth weight ranged from -1 to -4.7, for birth length from +0.1 to -4, and for head circumference from -1.4 to -5.
Postnatal growth. Poor weight gain was observed in all individuals who lived more than 24 hours. Z scores for weight at time of last evaluation ranged from -2.7 to -6.
Feeding problems in infancy with poor growth are common. Three individuals have required gastrostomy feeds for poor feeding and/or reflux with aspiration [Ehmke et al 2017; Author, unpublished data].
Short stature was present in nine of 11 individuals who lived more than 24 hours, showing poor linear growth with Z scores ranging from -1.2 to -6.
Microcephaly, proportionate to height, is frequently present.
Craniofacial Features
Turribrachycephaly or brachycephaly is apparent at birth in most individuals and may be noted prenatally. In all but two individuals for whom there are detailed infant records, large anterior fontanelle is present. In the 11 individuals who lived longer than the neonatal period, craniosynostosis (most often involving the coronal sutures) was confirmed in nine and suspected in another.
Skin and Hair
Excessively wrinkled, sagging, and thin skin is typically noted at birth. Dermal translucence with prominent vasculature and decreased subcutaneous fat is common. In combination these skin findings (reflecting an apparent lipodystrophy) contribute to a progeroid appearance in infancy that improves with time.
The hair pattern is distinctive. Hypertrichosis, most frequently over the back, neck, and face, is present at birth and persists at least through childhood. In neonates, sparse frontotemporal scalp hair with low anterior and posterior hairlines are frequently noted. With age, scalp hair becomes coarse and abnormal hair whorls and growth patterns are common (see Figure 1).
Skeletal
Digital abnormalities, present in all reported individuals, include a mix of short distal phalanges (11/13), cutaneous syndactyly (7/13), and nail aplasia or hypoplasia (12/13) [Ehmke et al 2017; Writzl et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Legué et al 2020; Author, unpublished data]. Radiographs show shortened distal phalanges.
Mixed nail hypoplasia and aplasia may be present on some digits; normal nails may be present on other digits. Postaxial digits of hands and feet tend to be more severely affected than preaxial digits.
Cutaneous syndactyly of hands or feet was present in seven individuals; in the six for whom it was specified, syndactyly involved toes 2-3 and 4-5 (in 2 individuals) [Ehmke et al 2017; Author, unpublished data], toes 2-3 only (1 individual), toes 4-5 only (1 individual) [Ehmke et al 2017, Legué et al 2020], fingers 3-4 only (1 individual), and fingers 4-5 only (1 individual) [Writzl et al 2017, Ryu et al 2019].
Other skeletal findings have been variably reported and include the following:
- Platyspondyly in two infants [Castori et al 2009; Author, unpublished]
- Nonspecific long bone diaphyseal and ossification abnormalities in one infant [Rodríguez et al 1999]
- Congenital hip dysplasia in one individual [Rodríguez-García et al 2018]
- Delayed bone age in one individual [Adolphs et al 2011]
Cardiovascular
Congenital heart defects (patent ductus arteriosus, atrial septal defect secundum, bicuspid aortic valve, and dysplastic valves) occurred in four individuals [Ehmke et al 2017, Writzl et al 2017].
Pulmonary hypertension occurred in four individuals during infancy [Ehmke et al 2017; Writzl et al 2017; Author, unpublished data].
Two had normal neonatal echocardiograms and developed pulmonary hypertension before age six months [Writzl et al 2017; Author, unpublished data].
Gastroesophageal reflux and aspiration requiring fundoplication and gastrostomy tube occurred in at least two individuals who also developed pulmonary hypertension [Ehmke et al 2017; Author, unpublished data]. In at least one individual, right heart pressure improved following gastric fundoplication [Author, unpublished data].
Aortic dilatation was seen in six of the 11 individuals who lived more than 24 hours. Aortic dilatation, which had not been present on neonatal echocardiograms, developed in at least three individuals who did not have other cardiac anomalies [Ehmke et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Legué et al 2020; Author, unpublished data]. In one individual dilatation progressed during treatment with atenolol [Rodríguez-García et al 2018]. Another individual presented with an ascending aortic dissection at age 45 years [Legué et al 2020].
Development
Delayed acquisition of milestones in infancy is common. Normal cognition has been described in individuals living beyond early childhood.
Specific details regarding acquisition of developmental milestones are limited to four individuals in whom independent sitting was achieved at 12-18 months, walking at 19-36 months, and speech at 16-24 months [Ehmke et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Author, unpublished data].
Other
Central nervous system
- Two individuals had hydrocephalus requiring shunt placement [Ehmke et al 2017]; these were the only individuals with relative macrocephaly compared to height.
- Two individuals had brain malformations: one with thin corpus callosum, ventriculomegaly, dysplastic cerebella vermis, pineal gland cyst, and large retrocerebellar area [Ehmke et al 2017]; and one with gyral simplification consistent with pachygyria, cerebellar hypoplasia, and subependymal heterotopia noted on autopsy [Writzl et al 2017].
- Five others had no abnormalities on brain imaging (MRI or CT) [Ehmke et al 2017; Writzl et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Author, unpublished data].
Ocular
- Hyperopia is common in individuals older than age one year [Ehmke et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Legué et al 2020; Author, unpublished data].
- Microphthalmia was present in several individuals [Rodríguez-García et al 2018, Ryu et al 2019, Legué et al 2020].
- One infant had iridocorneal adhesions and corneal clouding at birth [Writzl et al 2017].
Conductive hearing loss was present in six of eight individuals; at least one required myringotomy tubes for chronic middle ear effusion [Ehmke et al 2017; Writzl et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Legué et al 2020; Author, unpublished data].
Dentition
- Oligodontia, microdontia, and hypodontia have been reported.
- Primary dentition may be normal despite subsequent abnormalities of the permanent teeth [Ehmke et al 2017, Ryu et al 2019, Legué et al 2020].
Abdomen/gastrointestinal
- Umbilical hernia with diastasis recti or hypoplasia of abdominal musculature was present in 11 of 13 individuals [Ehmke et al 2017; Writzl et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Legué et al 2020; Author, unpublished data]. One individual had repair of umbilical hernia with subsequent recurrence [Ryu et al 2019].
- Intestinal malrotation was identified at autopsy in one infant [Writzl et al 2017].
Genital
- Hypoplastic labia were reported in 8 of 10 females [Ehmke et al 2017; Writzl et al 2017; Rodríguez-García et al 2018; Ryu et al 2019; Legué et al 2020; Author, unpublished data].
- Cryptorchidism was present in all three reported males. One also had micropenis [Writzl et al 2017] and one had hypospadias and cryptorchidism [Rodríguez-García et al 2018].
Severe and frequently life-limiting infections have been reported in six individuals [Ehmke et al 2017; Writzl et al 2017; Author, unpublished data]. In two of these individuals, persistent leukocytosis was also present [Writzl et al 2017; Author, unpublished data].
Prognosis
It is unknown whether life span in SLC25A24 Fontaine progeroid syndrome is abnormal. One individual with a molecularly confirmed diagnosis is alive at age 45 years, demonstrating that survival into adulthood is possible [Legué et al 2020].
Additionally, earlier reports of individuals clinically diagnosed with Gorlin-Chaudhry-Moss syndrome and Petty syndrome included individuals in their 30s and 40s [Petty et al 1990, Ippel et al 1992]. Since many adults with disabilities or malformations have not undergone advanced genetic testing, it is likely that adults with this condition are underrecognized and underreported.
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been identified. All individuals have a similar substitution at residue 217 (p.Arg217His or p.Arg217Cys) with no significant difference in clinical presentation (see Table 7).
Nomenclature
The following designations, previously thought to represent discrete phenotypes, were consolidated under the name "SLC25A24 Fontaine progeroid syndrome" following identification of their shared molecular etiology in 2017 [Ehmke et al 2017, Writzl et al 2017]:
- Hutchinson-Gilford progeria-like syndrome [Faivre et al 1999, Rodríguez et al 1999, Writzl et al 2017]
- Fontaine-Farriaux syndrome [Castori et al 2009, Writzl et al 2017]
- Gorlin-Chaudhry-Moss syndrome [Adolphs et al 2011, Ehmke et al 2017]
Although to date no individual with a clinical diagnosis of Petty syndrome has been confirmed to have a pathogenic variant in SLC25A24, the remarkably similar clinical features have led multiple authors to propose a shared etiology [Castori et al 2009, Braddock et al 2010, Writzl et al 2017, Ryu et al 2019].
Prevalence
The prevalence is unknown. To date, 13 individuals with molecularly confirmed SLC25A24 Fontaine progeroid syndrome have been reported worldwide.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in SLC25A24.
Differential Diagnosis
Table 3.
Genes of Interest in the Differential Diagnosis of SLC25A24 Fontaine Progeroid Syndrome
Management
No clinical practice guidelines for SLC25A24 Fontaine progeroid syndrome have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with SLC25A24 Fontaine progeroid syndrome, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with SLC25A24 Fontaine Progeroid Syndrome
Treatment of Manifestations
Management, which is largely symptomatic, may be performed by specialists in multiple disciplines, including a craniofacial clinic (involving plastic surgery, neurosurgery, and otolaryngology), cardiology, pulmonology, gastroenterology, and clinical genetics. Some students may benefit from an individualized education plan (IEP) through their school.
Table 5.
Treatment of Manifestations in Individuals with SLC25A24 Fontaine Progeroid Syndrome
Developmental Delay Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
- For muscle tone abnormalities including hypertonia or dystonia, consider involving appropriate specialists to aid in management of baclofen, tizanidine, Botox®, anti-parkinsonian medications, or orthopedic procedures.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Surveillance
Table 6.
Recommended Surveillance for Individuals with SLC25A24 Fontaine Progeroid Syndrome
Agents/Circumstances to Avoid
Contact sports and isometric exercise may need to be restricted if cranial anomalies and/or aortic dilatation are present. Note, in the absence of severe aortic dilatation or other clinical restriction, aerobic activity is encouraged.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
SLC25A24 Fontaine progeroid syndrome is an autosomal dominant disorder typically caused by a de novo pathogenic variant. All probands reported to date whose parents have undergone molecular genetic testing have the disorder as a result of a de novo SLC25A24 pathogenic variant.
Risk to Family Members
Parents of a proband
- All probands reported to date with SLC25A24 Fontaine progeroid syndrome whose parents have undergone molecular genetic testing have the disorder as a result of a de novo SLC25A24 pathogenic variant.
- Molecular genetic testing is recommended for the parents of the proband to confirm their genetic status and to allow reliable recurrence risk counseling.
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present only in the germ cells.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If a parent of the proband is known to have the SLC25A24 pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%.
- If the SLC25A24 pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
Offspring of a proband. Individuals with SLC25A24 Fontaine progeroid syndrome are not known to reproduce and fertility has not been assessed.
Other family members. Given that all probands with SLC25A24 Fontaine progeroid syndrome reported to date have the disorder as a result of a de novo SLC25A24 pathogenic variant, the risk to other family members is presumed to be low.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to parents of affected individuals.
Prenatal Testing and Preimplantation Genetic Testing
Risk to future pregnancies is presumed to be low as the proband most likely has a de novo SLC25A24 pathogenic variant. There is, however, a recurrence risk (~1%) to sibs based on the theoretic possibility of parental germline mosaicism [Rahbari et al 2016]. Given this risk, prenatal and preimplantation genetic testing may be considered.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Children's Craniofacial AssociationPhone: 800-535-3643Email: contactCCA@ccakids.com
- FACES: National Craniofacial AssociationPhone: 800-332-2373; 423-266-1632Email: info@faces-cranio.org
- National Organization for Rare Disorders (NORD)Phone: 800-999-6673
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
SLC25A24 Fontaine Progeroid Syndrome: Genes and Databases
Table B.
OMIM Entries for SLC25A24 Fontaine Progeroid Syndrome (View All in OMIM)
Molecular Pathogenesis
SLC25A24 encodes calcium-binding mitochondrial carrier protein SCaMC-1. A calcium-sensitive N-domain regulatory region that is followed by six transmembrane helices forms an inner mitochondrial transmembrane domain that (1) regulates the intramembranous adenine pool via transporting adenine nucleotides in exchange for phosphate [Palmieri et al 2020] and (2) may play a role in resistance to oxidative stress by contributing to formation of the mitochondrial permeability transition pore [Ehmke et al 2017].
Knockdown of SLC25A24 contributes to the oxidative stress and calcium overload and increases susceptibility to mitochondrial permeability transition pore-dependent cell death [Traba et al 2012].
Mechanism of disease causation. The specific mechanism by which SLC25A24 variants result in the multisystem clinical features of SLC25A24 Fontaine progeroid syndrome is unclear.
Pathogenic variants causative of Fontaine progeroid syndrome have been restricted to codon 217 in exon 5 of SLC25A24. This position is in the transmembrane domain, located at the end of the predicted helix 1. The Arg217 residue is part of the conserved mitochondrial carrier family motif, with substitutions to this residue expected to narrow the substrate cavity [Writzl et al 2017]. Mitochondrial fragmentation and swelling in response to oxidative stress in cells expressing missense variants supports a gain-of-function effect, interfering with regulation of the mitochondrial permeability transition pore [Ehmke et al 2017].
SLC25A24-specific laboratory technical considerations. Pathogenic variants causative of Fontaine progeroid syndrome have been restricted to codon 217 in exon 5 of SLC25A24. See Table 7.
Table 7.
Notable SLC25A24 Pathogenic Variants
Chapter Notes
Revision History
- 9 June 2022 (bp) Review posted live
- 1 March 2022 (dv) Original submission
References
Literature Cited
- Adolphs N, Klein M, Haberl EJ, Graul-Neumann L, Menneking H, Hoffmeister B. Necrotizing soft tissue infection of the scalp after fronto-facial advancement by internal distraction in a 7-year old girl with Gorlin-Chaudhry-Moss syndrome--a case report. J Craniomaxillofac Surg. 2011;39:554–61. [PubMed: 21216154]
- Braddock SR, Ardinger HH, Yang CS, Paschal BM, Hall BD. Petty syndrome and Fontaine-Farriaux syndrome: delineation of a single syndrome. Am J Med Genet A. 2010;152A:1718–23. [PubMed: 20583180]
- Castori M, Silvestri E, Pedace L, Marseglia G, Tempera A, Antigoni I, Torricelli F, Majore S, Grammatico P. Fontaine-Farriaux syndrome: a recognizable craniosynostosis syndrome with nail, skeletal, abdominal, and central nervous system anomalies. Am J Med Genet A. 2009;149A:2193–9. [PubMed: 19731360]
- Ehmke N, Graul-Neumann L, Smorag L, Koenig R, Segebrecht L, Magoulas P, Scaglia F, Kilic E, Hennig AF, Adolphs N, Saha N, Fauler B, Kalscheuer VM, Hennig F, Altmuller J, Netzer C, Thiele H, Nurnberg P, Yigit G, Jager M, Hecht J, Kruger U, Mielke T, Krawitz PM, Horn D, Schuelke M, Mundlos S, Bacino CA, Bonnen PE, Wollnik B, Fischer-Zirnsak B, Kornak U. De novo mutations in SLC25A24 cause a craniosynostosis syndrome with hypertrichosis, progeroid appearance, and mitochondrial dysfunction. Am J Hum Genet. 2017;101:833–43. [PMC free article: PMC5673623] [PubMed: 29100093]
- Faivre L, Khau Van Kien P, Madinier-Chappat N, Nivelon-Chevallier A, Beer F, LeMerrer M. Can Hutchinson-Gilford progeria syndrome be a neonatal condition? Am J Med Genet. 1999;87:450–2. [PubMed: 10594888]
- Fontaine G, Farriaux JP, Blanckaert D, Lefebvre C. Un nouveau syndrome polymalformatif complexe. J Genet Hum. 1977;25:109–19. [PubMed: 21227]
- Gorlin RJ, Chaudhry AP, Moss ML. Craniofacial dysostosis, patent ductus arteriosus, hypertrichosis, hypoplasia of labia majora, dental and eye anomalies-a new syndrome? J Pediatr. 1960;56:778–85. [PubMed: 13851313]
- Ippel PF, Gorlin RJ, Lenz W, van Doorne JM, Bijlsma JB. Craniofacial dysostosis, hypertrichosis, genital hypoplasia, ocular, dental, and digital defects: confirmation of the Gorlin-Chaudhry-Moss syndrome. Am J Med Genet. 1992;44:518–22. [PubMed: 1442899]
- Legué J, François JHM, van Rijswijk CSP, van Brakel TJ. Is Gorlin-Chaudhry-Moss syndrome associated with aortopathy? Eur J Cardiothorac Surg. 2020;58:654–5. [PMC free article: PMC7453031] [PubMed: 32355952]
- Palmieri F, Scarcia P, Monné M. Diseases caused by mutations in mitochondrial carrier genes SLC25: a review. Biomolecules. 2020;10:655. [PMC free article: PMC7226361] [PubMed: 32340404]
- Petty EM, Laxova R, Wiedemann HR. Previously unrecognized congenital progeroid disorder. Am J Med Genet. 1990;35:383–7. [PubMed: 2309786]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Rodríguez JI, Pérez-Alonso P, Funes R, Pérez-Rodríguez J. Lethal neonatal Hutchinson-Gilford progeria syndrome. Am J Med Genet. 1999;82:242–8. [PubMed: 10215548]
- Rodríguez-García ME, Cotrina-Vinagre FJ, Cruz-Rojo J, Garzón-Lorenzo L, Carnicero-Rodríguez P, Pozo JS, Martínez-Azorín F. A rare male patient with Fontaine progeroid syndrome caused by p.R217H de novo mutation in SLC25A24. Am J Med Genet A. 2018;176:2479–86. [PubMed: 30329211]
- Ryu J, Ko JM, Shin CH. A. 9-year-old Korean girl with Fontaine progeroid syndrome: a case report with further phenotypical delineation and description of clinical course during long-term follow-up. BMC Med Genet. 2019;20:188. [PMC free article: PMC6882017] [PubMed: 31775791]
- Traba J, Del Arco A, Duchen MR, Szabadkai G, Satrústegui J. SCaMC-1 promotes cancer cell survival by desensitizing mitochondrial permeability transition via ATP/ADP-mediated matrix Ca(2+) buffering. Cell Death Differ. 2012;19:650–60. [PMC free article: PMC3307981] [PubMed: 22015608]
- Writzl K, Maver A, Kovacic L, Martinez-Valero P, Contreras L, Satrustegui J, Castori M, Faivre L, Lapunzina P, van Kuilenburg ABP, Radovic S, Thauvin-Robinet C, Peterlin B, Del Arco A, Hennekam RC. De novo mutations in SLC25A24 cause a disorder characterized by early aging, bone dysplasia, characteristic face, and early demise. Am J Hum Genet. 2017;2017;101:844–55. [PMC free article: PMC5673633] [PubMed: 29100094]
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: June 9, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Velasco D, Olney AH, Starr L. SLC25A24 Fontaine Progeroid Syndrome. 2022 Jun 9. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.


