Summary
Clinical characteristics.
THOC6 intellectual disability syndrome is associated with moderate-to-severe developmental delay or intellectual disability; nonspecific dysmorphic facial features (tall forehead, deep-set eyes, short and upslanted palpebral fissures, epicanthal folds, and long nose with low-hanging columella); microcephaly (typically 2-3 SD below the mean); teeth anomalies (dental caries, malocclusion, and supernumerary teeth); cardiac anomalies (most typically atrial and/or ventricular septal defects); prenatal ventriculomegaly and hydrocephalus; cryptorchidism in males; and renal malformations (most commonly unilateral renal agenesis). More rarely, affected individuals may have hypergonadotropic hypogonadism (in females), seizures, poor growth, feeding difficulties, hearing loss, refractive errors and/or other eye abnormalities, vertebral anomalies, micro/retrognathia, and imperforate / anteriorly placed anus.
Diagnosis/testing.
The diagnosis of THOC6 intellectual disability syndrome is established in a proband with biallelic pathogenic variants in THOC6 identified by molecular genetic testing. For individuals from the Hutterite population suspected of having THOC6 intellectual disability syndrome, molecular genetic testing for the specific c.136G>A (p.Gly46Arg) founder variant can be considered.
Management.
Treatment of manifestations: For those with poor weight gain, feeding therapy and consideration of a gastrostomy tube; for those with hearing loss, hearing aids may be considered; standard treatment for seizures, vision issues, dental caries/malocclusion, cardiac malformations, genital anomalies, hypergonadotropic hypogonadism, renal malformations, skeletal anomalies, and developmental delay / intellectual disability.
Surveillance: At each visit: monitor developmental progress, mobility, self-help skills, and behavior; assess for signs and symptoms of hydrocephalus or for new neurologic manifestations; measurement of growth parameters and evaluation of nutritional status; assessment of vision and eye alignment; assessment for dental caries and malocclusion. Evaluate renal function (BUN, creatinine, and urinalysis) at each visit or annually for those with anomalies of the kidney and urinary tract; annual audiology evaluation; evaluation of secondary sexual characteristics and menstrual cycles at each visit in females older than age 12 years.
Genetic counseling.
THOC6 intellectual disability syndrome is inherited in an autosomal recessive manner. At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being unaffected and a carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk relatives and prenatal testing for pregnancies at increased risk are possible if the pathogenic variants have been identified in an affected family member.
Diagnosis
Formal diagnostic criteria for THOC6 intellectual disability syndrome have not been established.
Suggestive Findings
THOC6 intellectual disability syndrome should be considered in individuals with the following clinical and imaging findings:
- Moderate-to-severe developmental delay (DD) or intellectual disability (ID)AND
- One or more of the following features presenting in infancy or childhood:
- Microcephaly
- Multiple dental caries and/or dental malocclusion
- Nonspecific dysmorphic features, including tall forehead, deep set eyes, short and upslanted palpebral fissures, epicanthal folds and long nose with low hanging columella
- Cryptorchidism in males
- Structural cardiac anomalies
- Structural renal anomalies
- Ventriculomegaly on brain imaging
Establishing the Diagnosis
The diagnosis of THOC6 intellectual disability syndrome is established in a proband with biallelic pathogenic (or likely pathogenic) variants in THOC6 identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic THOC6 variants of uncertain significance (or of one known THOC6 pathogenic variant and one THOC6 variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular Genetic Testing
Molecular genetic testing in a child with developmental delay or an older individual with intellectual disability typically begins with chromosomal microarray analysis (CMA). CMA uses oligonucleotide and/or SNP arrays to detect genome-wide large deletions/duplications (including THOC6) that cannot be detected by sequence analysis.
If CMA is not diagnostic, the next step is typically either a multigene panel or exome sequencing.
- For individuals from the Hutterite population suspected of having THOC6 intellectual disability syndrome, molecular genetic testing for the specific c.136G>A (p.Gly46Arg) founder variant can be considered first (see Molecular Genetics).
- For individuals who do not originate from the Hutterite population, single-gene testing (sequence analysis of THOC6, followed by gene-targeted deletion/duplication analysis) is rarely useful and typically NOT recommended.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (CMA, exome sequencing, exome array, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of THOC6 intellectual disability syndrome is somewhat nonspecific, the majority of affected individuals have a phenotype indistinguishable from many other inherited disorders with intellectual disability. Therefore, targeted genomic testing (Option 1) or comprehensive genomic testing (Option 2) are the most reasonable methods to detect this condition.
Option 1
An intellectual disability or microcephaly multigene panel that includes THOC6 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition in a person with a nondiagnostic CMA while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. Of note, given the rarity of THOC6 Intellectual disability syndrome, some panels for intellectual disability may not include this gene. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by intellectual disability and other malformations, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in THOC6 Intellectual Disability Syndrome
Clinical Characteristics
Clinical Description
THOC6 intellectual disability syndrome is associated with intellectual disability, distinctive facial features, microcephaly, teeth anomalies, and cardiac and renal malformations. To date, 19 individuals from 15 families with pathogenic variants in THOC6 have been identified [Boycott et al 2010, Beaulieu et al 2013, Anazi et al 2016, Casey et al 2016, Amos et al 2017, Accogli et al 2018, Nair et al 2018, Bruel et al 2019, Elmas et al 2019, Mattioli et al 2019, Gupta et al 2020, Zhang et al 2020]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Features of THOC6 Intellectual Disability Syndrome
Developmental Delay (DD) / Intellectual Disability (ID)
Moderate-to-severe intellectual disability has been noted in all reported individuals. Most of these individuals were able to walk independently, but remained nonverbal or had very limited speech (<10 words). The oldest reported individuals are young adults.
Behavior problems. Autism spectrum disorder and motor stereotypies were described in four individuals [Accogli et al 2018, Elmas et al 2019, Mattioli et al 2019]. Obsessive compulsive behavior was observed in one individual [Amos et al 2017].
Neurologic
Most reported individuals had congenital microcephaly, predominantly 2-3 SD below the mean; 5 SD below the mean was observed in one child [Accogli et al 2018].
Epilepsy. Two affected individuals were reported to have seizures; seizure type and severity was not specified [Elmas et al 2019, Mattioli et al 2019].
Neuroimaging. Ventriculomegaly was reported prenatally in four individuals [Casey et al 2016, Accogli et al 2018, Elmas et al 2019, Mattioli et al 2019] and in six affected individuals in total [Amos et al 2017, Nair et al 2018].
- One child had postnatal hydrocephalus that required ventriculoperitoneal shunt placement [Mattioli et al 2019].
- One individual had compensated supratentorial hydrocephalus due to aqueductal stenosis and was also reported to have cerebellar hypoplasia with severe vermian dysgenesis, small pons, hippocampal dysgenesis, and partial agenesis of the septum pellucidum [Accogli et al 2018].
Corpus callosum dysgenesis was identified in five reported individuals [Amos et al 2017, Mattioli et al 2019, Bruel et al 2019, Elmas et al 2019].
Growth
Low birth weight was present in most reported individuals, and intrauterine growth restriction was documented in four [Casey et al 2016, Amos et al 2017, Accogli et al 2018, Mattioli et al 2019].
Five individuals had failure to thrive in childhood [Anazi et al 2016, Accogli et al 2018, Gupta et al 2020, Zhang et al 2020].
Eight reported individuals were of short stature, in the range of 2-3 SD below the mean.
Gastrointestinal Problems
Three individuals presented with feeding difficulties, two requiring feeding through a gastrostomy tube because of inadequate caloric intake by mouth [Casey et al 2016, Mattioli et al 2019]. Three individuals had anal anomalies, including anal atresia, anteriorly positioned anus, and a rectoperineal fistula [Anazi et al 2016, Amos et al 2017, Accogli et al 2018]. One affected child was reported to have had a mesenteric cyst that required surgical correction [Zhang et al 2020].
Facial Features
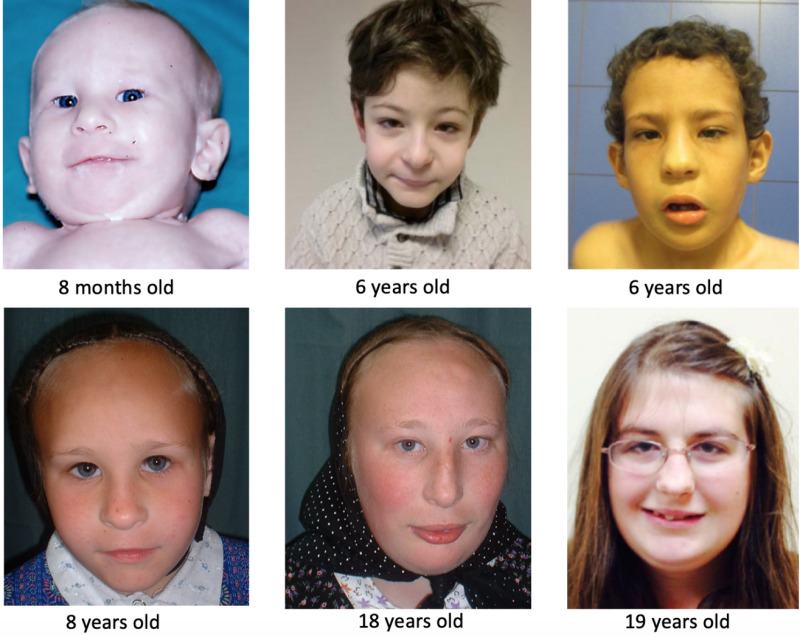
Dysmorphic facial features were present in most reported individuals and included the following: tall forehead, deep-set eyes, short and upslanted palpebral fissures, epicanthal folds, and long nose with low-hanging columella (Figure 1). Facial features are typically not striking enough to allow a clinician to recognize the condition on this basis alone; however, the features are often recognized as consistent with this diagnosis after the diagnosis has been suggested or confirmed.

Sensory Impairment
Hearing loss was reported in three affected individuals, and was specified to be of sensorineural origin in one [Amos et al 2017, Mattioli et al 2019].
Eyes. Myopia was noted in three individuals [Boycott et al 2010, Gupta et al 2020].
Other reported ocular anomalies include bilateral optic disc hypoplasia [Accogli et al 2018] and alternating exotropia, nystagmus, and hyperopia [Mattioli et al 2019].
ENT/Mouth
Micro-/retrognathia, reported in five individuals, was not significant enough to cause respiratory impairment [Boycott et al 2010, Amos et al 2017, Mattioli et al 2019].
Palatal anomalies. A cleft palate and a submucous cleft palate were seen in two individuals; one of them also had choanal atresia [Amos et al 2017, Mattioli et al 2019]. A bifid uvula and velopharyngeal insufficiency were reported in one individual each [Boycott et al 2010, Accogli et al 2018].
Teeth anomalies. Multiple dental caries and/or dental malocclusion were observed in ten affected individuals. Supernumerary teeth were also reported in one child [Accogli et al 2018].
Cardiovascular Anomalies
Atrial septal defect, ventricular septal defect, and patent ductus arteriosus were present in eight reported individuals. A dysmorphic and mildly insufficient mitral valve was also seen in one individual [Accogli et al 2018], and pulmonary hypertension in another [Mattioli et al 2019].
Genital Anomalies / Puberty
Males
- Cryptorchidism, bilateral or unilateral, was present in seven affected males.
- Two males presented with a micropenis [Nair et al 2018, Mattioli et al 2019], and one of them also had a hypospadias [Mattioli et al 2019].
Females
- Premature ovarian failure was identified in one teenage girl [Boycott et al 2010].
- Hypergonadotropic hypogonadism with primary amenorrhea requiring hormone replacement therapy was reported in another girl [Accogli et al 2018].
- Endometriosis was reported in one female [Boycott et al 2010].
Renal Anomalies
Unilateral renal agenesis was identified in four individuals [Beaulieu et al 2013, Casey et al 2016, Gupta et al 2020]. One of the individuals with unilateral renal agenesis also had a contralateral echogenic and atrophic kidney. She developed renal failure requiring dialysis at age 13 years and underwent kidney transplantation at age 15 years [Beaulieu et al 2013].
An ectopic kidney located in the pelvis or a horseshoe kidney was reported in three individuals [Beaulieu et al 2013, Accogli et al 2018, Gupta et al 2020].
Recurrent urinary tract infections were seen in three individuals [Boycott et al 2010, Amos et al 2017].
Skeletal Features
Cervical hemivertebrae and multilevel vertebral segmentation defects causing a scoliosis were reported in two different individuals [Accogli et al 2018, Gupta et al 2020].
Two individuals presented with camptodactyly [Anazi et al 2016, Accogli et al 2018].
Other reported anomalies include pes planus, trigger thumb, calcaneovalgus and equinovarus deformities, cubitus valgus, congenital hip dislocation, radioulnar joint dysostosis, cervical rib, and Sprengel deformity.
Prognosis
It is unknown whether life span in THOC6 intellectual disability syndrome is shortened. The original four affected individuals are now young adults, with the oldest in their early 40s [Author, personal communication], demonstrating that survival into adulthood is possible. Since many adults with disabilities have not undergone advanced genetic testing, it is likely that adults with this condition are underrecognized and underreported.
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been identified.
Prevalence
As of early 2020, seven years after this syndrome was first described and molecularly explained, 19 affected individuals have been reported in the literature.
Initially reported in the Hutterite population [Boycott et al 2010], THOC6 intellectual disability syndrome has now been identified in individuals worldwide. This disorder was more prevalent in two Hutterite leuts, with a specific founder variant (c.136G>A;p.Gly46Arg) frequency of 3% in Dariusleut controls and of 2% in Lehrerleut controls (see Molecular Genetics) [Beaulieu et al 2013].
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in THOC6.
Differential Diagnosis
Several intellectual disability disorders are associated with additional features that overlap those observed in THOC6 intellectual disability syndrome (see Table 3).
However, because the phenotypic features associated with THOC6 intellectual disability syndrome can be nonspecific, all disorders with intellectual disability without other distinctive findings should be considered in the differential diagnosis. See OMIM Autosomal Dominant, Autosomal Recessive, Nonsyndromic X-Linked, and Syndromic X-Linked Intellectual Developmental Disorder Phenotypic Series.
Table 3.
Genes of Interest in the Differential Diagnosis of THOC6 Intellectual Disability Syndrome
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with THOC6 intellectual disability syndrome, the evaluations summarized in this section (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with THOC6 Intellectual Disability Syndrome
Treatment of Manifestations
Table 5.
Treatment of Manifestations in Individuals with THOC6 Intellectual Disability Syndrome
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies, and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures, scoliosis, hip dislocation).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
- For muscle tone abnormalities including hypertonia or dystonia, consider involving appropriate specialists to aid in management of baclofen, tizanidine, Botox®, antiparkinsonian medications, or orthopedic procedures.
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. When feeding dysfunction is severe, an NG-tube or G-tube may be necessary.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Social/Behavioral Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder, when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
Table 6.
Recommended Surveillance for Individuals with THOC6 Intellectual Disability Syndrome
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
THOC6 intellectual disability syndrome is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., presumed to be carriers of one THOC6 pathogenic variant based on family history).
- Rarely, only one parent is heterozygous for a THOC6 pathogenic variant and the child has the disorder as the result of uniparental isodisomy for chromosome 16 and consequent homozygosity for the THOC6 pathogenic variant from the carrier parent (reported once previously by Mattioli et al [2019]).
- Accurate recurrence risk counseling relies on carrier testing of both parents to determine if each is heterozygous for a THOC6 variant. If carrier testing detects the variant in only one parent:
- And the child appears to have homozygous THOC6 pathogenic variants, possible explanations include a large deletion on one allele (if not previously tested for) and uniparental isodisomy for chromosome 16;
- And the child has compound heterozygous THOC6 pathogenic variants, the child may theoretically have one inherited variant and one de novo pathogenic variant (de novo variants are known to occur at a low but appreciable rate in autosomal recessive disorders [Jónsson et al 2017]).
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for a THOC6 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- If the proband has the disorder as the result of uniparental isodisomy for chromosome 16 and only one parent is heterozygous for a THOC6 pathogenic variant, each sib of an affected individual has at conception a 50% chance of being an asymptomatic carrier and a 50% chance of being unaffected and not a carrier. (The risk to the sibs of being affected with THOC6 intellectual disability syndrome is not increased over that of the general population.)
Offspring of a proband. To date, individuals with THOC6 intellectual disability syndrome are not known to reproduce.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is heterozygous for an THOC6 pathogenic variant, the parent's family members are at risk of being a carrier.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the THOC6 pathogenic variants in the family.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the THOC6 pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- American Association on Intellectual and Developmental Disabilities (AAIDD)Phone: 202-387-1968
- MedlinePlus
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
THOC6 Intellectual Disability Syndrome: Genes and Databases
Table B.
OMIM Entries for THOC6 Intellectual Disability Syndrome (View All in OMIM)
Molecular Pathogenesis
THOC6 encodes a member of the THO complex, which is composed of multiple subunits: THOC1, THOC2, THOC3, THOC5, THOC6, and THOC7 [Masuda et al 2005, Guria et al 2011]. The THO complex is part of a larger TREX (transcription/export) complex, which is involved in the processing of mRNA transcription, as well as the export of spliced mRNA from the nucleus [Masuda et al 2005]. Of note, the THO complex has been shown to be involved in stem cell renewal and to be an important regulator of neuronal differentiation, brain synapse development, and dopamine neuron survival [Wang et al 2013, Maeder et al 2018].
Mechanism of disease causation. Loss of function. The p.Gly46Arg variant causes mislocalization of THOC6 in the cytoplasm [Beaulieu et al 2013]. The triple variant haplotype (p.Trp100Arg; p.Val234Leu; p.Gly275Asp) also results in mislocalization of THOC6 into the cytoplasm and a decrease in its ability to interact with other members of the THO complex [Mattioli et al 2019].
Table 7.
Notable THOC6 Pathogenic Variants
Chapter Notes
Author Notes
Of note, there is an online RareConnect community for THOC6 intellectual disability syndrome (Beaulieu-Boycott-Innes syndrome) that connects affected families. See www.rareconnect.org/en/community/bbis.
Revision History
- 13 August 2020 (ma) Review posted live
- 18 March 2020 (kb) Original submission
References
Literature Cited
- Accogli A, Scala M, Calcagno A, Castello R, Torella A, Musacchia F, Allegri AME, Mancardi MM, Maghnie M, Severino M, Nigro V, Capra V, et al. Novel CNS malformations and skeletal anomalies in a patient with Beaulieu-Boycott-Innes syndrome. Am J Med Genet A. 2018;176:2835–40. [PubMed: 30238602]
- Amos JS, Huang L, Thevenon J, Kariminedjad A, Beaulieu CL, Masurel-Paulet A, Najmabadi H, Fattahi Z, Beheshtian M, Tonekaboni SH, Tang S, Helbig KL, Alcaraz W, Riviere JB, Faivre L, Innes AM, Lebel RR, Boycott KM, et al. Autosomal recessive mutations in THOC6 cause intellectual disability: syndrome delineation requiring forward and reverse phenotyping. Clin Genet. 2017;91:92–9. [PubMed: 27102954]
- Anazi S, Alshammari M, Moneis D, Abouelhoda M, Ibrahim N, Alkuraya FS. Confirming the candidacy of THOC6 in the etiology of intellectual disability. Am J Med Genet A. 2016;170A:1367–9. [PubMed: 26739162]
- Beaulieu CL, Huang L, Innes AM, Akimenko MA, Puffenberger EG, Schwartz C, Jerry P, Ober C, Hegele RA, Mcleod DR, Schwartzentruber J, Majewski J, Bulman DE, Parboosingh JS, Boycott KM, et al. Intellectual disability associated with a homozygous missense mutation in THOC6. Orphanet J Rare Dis. 2013;8:62. [PMC free article: PMC3644499] [PubMed: 23621916]
- Boycott KM, Beaulieu C, Puffenberger EG, Mcleod DR, Parboosingh JS, Innes AM. A novel autosomal recessive malformation syndrome associated with developmental delay and distinctive facies maps to 16ptel in the Hutterite population. Am J Med Genet A. 2010;152A:1349–56. [PubMed: 20503307]
- Bruel AL, Nambot S, Quere V, Vitobello A, Thevenon J, Assoum M, Moutton S, Houcinat N, Lehalle D, Jean-Marcais N, Chevarin M, Jouan T, Poe C, Callier P, Tisserand E, Philippe C, Them FTM, Duffourd Y, Faivre L, Thauvin-Robinet C, et al. Increased diagnostic and new genes identification outcome using research reanalysis of singleton exome sequencing. Eur J Hum Genet. 2019;27:1519–31. [PMC free article: PMC6777617] [PubMed: 31231135]
- Casey J, Jenkinson A, Magee A, Ennis S, Monavari A, Green A, Lynch SA, Crushell E, Hughes J. Beaulieu-Boycott-Innes syndrome: an intellectual disability syndrome with characteristic facies. Clin Dysmorphol. 2016;25:146–51. [PubMed: 27295358]
- Elmas M, Yildiz H, Erdogan M, Gogus B, Avci K, Solak M. Comparison of clinical parameters with whole exome sequencing analysis results of autosomal recessive patients; a center experience. Mol Biol Rep. 2019;46:287–99. [PubMed: 30426380]
- Gupta N, Yadav S, Gurramkonda VB, Ramprasad VL, Thenral SG, Kabra M. First report of THOC6 related intellectual disability (Beaulieu-Boycott-Innes syndrome) in two siblings from India. Eur J Med Genet. 2020;63:103742. [PubMed: 31421288]
- Guria A, Tran DD, Ramachandran S, Koch A, El Bounkari O, Dutta P, Hauser H, Tamura T. Identification of mRNAs that are spliced but not exported to the cytoplasm in the absence of THOC5 in mouse embryo fibroblasts. RNA. 2011;17:1048–56. [PMC free article: PMC3096037] [PubMed: 21525145]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Maeder CI, Kim JI, Liang X, Kaganovsky K, Shen A, Li Q, Li Z, Wang S, Xu XZS, Li JB, Xiang YK, Ding JB, Shen K. The THO complex coordinates transcripts for synapse development and dopamine neuron survival. Cell. 2018;174:1436–49.e20. [PMC free article: PMC6542560] [PubMed: 30146163]
- Masuda S, Das R, Cheng H, Hurt E, Dorman N, Reed R. Recruitment of the human TREX complex to mRNA during splicing. Genes Dev. 2005;19:1512–7. [PMC free article: PMC1172058] [PubMed: 15998806]
- Mattioli F, Isidor B, Abdul-Rahman O, Gunter A, Huang L, Kumar R, Beaulieu C, Gecz J, Innes M, Mandel JL, Piton A. Clinical and functional characterization of recurrent missense variants implicated in THOC6-related intellectual disability. Hum Mol Genet. 2019;28:952–60. [PubMed: 30476144]
- Nair P, Sabbagh S, Mansour H, Fawaz A, Hmaimess G, Noun P, Dagher R, Megarbane H, Hana S, Alame S, Lamaa M, Hasbini D, Farah R, Rajab M, Stora S, El-Tourjuman O, Abou Jaoude P, Chalouhi G, Sayad R, Gillart AC, Al-Ali M, Delague V, El-Hayek S, Megarbane A. Contribution of next generation sequencing in pediatric practice in Lebanon. A study on 213 cases. Mol Genet Genomic Med. 2018;6:1041–52. [PMC free article: PMC6305638] [PubMed: 30293248]
- Wang L, Miao YL, Zheng X, Lackford B, Zhou B, Han L, Yao C, Ward JM, Burkholder A, Lipchina I, Fargo DC, Hochedlinger K, Shi Y, Williams CJ, Hu G. The THO complex regulates pluripotency gene mRNA export and controls embryonic stem cell self-renewal and somatic cell reprogramming. Cell Stem Cell. 2013;13:676–90. [PMC free article: PMC3962795] [PubMed: 24315442]
- Zhang Q, Chen S, Qin Z, Zheng H, Fan X. The first reported case of Beaulieu-Boycott-Innes syndrome caused by two novel mutations in THOC6 gene in a Chinese infant. Medicine (Baltimore). 2020;99:e19751. [PMC free article: PMC7220430] [PubMed: 32282736]
Publication Details
Author Information and Affiliations
University of Ottawa
Ottawa, Canada
Alberta Children's Hospital Research Institute
University of Calgary
Calgary, Canada
Children's Hospital of Eastern Ontario
University of Ottawa
Ottawa, Canada
Publication History
Initial Posting: August 13, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Lemire G, Innes AM, Boycott KM. THOC6 Intellectual Disability Syndrome. 2020 Aug 13. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.