Summary
Clinical characteristics.
Chediak-Higashi syndrome (CHS) is characterized by partial oculocutaneous albinism (OCA), immunodeficiency, a mild bleeding tendency, and late adolescent- to adult-onset neurologic manifestations (e.g., learning difficulties, peripheral neuropathy, ataxia, and parkinsonism). While present in nearly all individuals with CHS, these clinical findings vary in severity.
Of note, all individuals with CHS are at risk of developing neurologic manifestations and hemophagocytic lymphohistiocytosis (HLH).
Individuals with severe childhood-onset presentations are considered to have "classic" CHS, whereas individuals with milder adolescent- to adult-onset presentations are considered to have "atypical" CHS. Because of the considerable overlap between classic CHS and atypical CHS, the disorder is best understood as a continuum of severe to milder phenotypes, with the universal feature being the pathognomonic giant granules within leukocytes observed on peripheral blood smear.
Diagnosis/testing.
The clinical diagnosis of CHS is established in a proband with suggestive clinical findings by identification of the pathognomonic giant granules within leukocytes on peripheral blood smear and/or biallelic pathogenic variants in LYST on molecular genetic testing.
Management.
Targeted therapy: The only targeted therapy currently available is hematopoietic stem cell transplantation (HSCT). HSCT can correct the hematologic and immunologic manifestations of CHS but does not appear to protect against the development of neurologic manifestations.
Supportive care: Multidisciplinary care is recommended, including specialists in ophthalmology and low vision services, infectious disease for management and prevention, hematology (to manage the bleeding disorder, HSCT, and treatment of HLH), neurology and physiatry, physical therapy, occupational therapy, and (for children) neuropsychology or developmental pediatrics to address educational and emotional needs or (for adults) neuropsychology.
Surveillance: To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, routinely scheduled follow-up evaluations by the multidisciplinary specialists are recommended.
Agents/circumstances to avoid: Live vaccines given the risk of infection due to immunodeficiency; all nonsteroidal anti-inflammatory drugs (NSAIDs) (e.g., aspirin, ibuprofen) given the risk of exacerbating the bleeding tendency.
Evaluation of relatives at risk: It is appropriate to evaluate the older and younger sibs of a proband as early as possible. Early diagnosis may provide the opportunity to perform HSCT prior to the development of HLH.
Pregnancy management: Although data are limited, to date females with CHS report uneventful pregnancy, labor, and delivery. However, because of concerns about bleeding during delivery and the postpartum period, developing a plan prior to delivery to address this issue is recommended.
Genetic counseling.
CHS is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a LYST pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the LYST pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives and prenatal/preimplantation genetic testing are possible.
GeneReview Scope
Chediak-Higashi syndrome (CHS) is characterized by partial oculocutaneous albinism (OCA), immunodeficiency, a mild bleeding tendency, and late adolescent- to adult-onset neurologic manifestations (e.g., learning difficulties, peripheral neuropathy, ataxia, and parkinsonism). All individuals are at risk of developing hemophagocytic lymphohistiocytosis (HLH).
While present in nearly all individuals with CHS, these clinical findings vary in severity. Individuals with severe presentations (see GeneReview Scope) are considered to have "classic" CHS, whereas individuals with milder presentations are considered to have "atypical" CHS. Because of the considerable overlap between classic CHS and atypical CHS, the disorder is best understood as a continuum of severe to milder phenotypes, with the universal feature being the pathognomonic giant granules within leukocytes observed on peripheral blood smear.
Table
GeneReview Scope: Chediak-Higashi Syndrome Clinical Continuum
Diagnosis
Suggestive Findings
The diagnosis of Chediak-Higashi syndrome (CHS) should be suspected in a proband with any of the following clinical features, supportive laboratory findings, and family history.
Clinical features
- Oculocutaneous albinism (OCA) with residual pigmentation characterized by:
- Signs and symptoms of low vision typical of OCA
- Reduced iris pigmentation (manifesting as iris transillumination often only on ophthalmologic examination). Note that irides may be darker than the light blue that is often associated with OCA.
- Reduced retinal pigmentation
- Hair that may have a silvery-gray sheen
- A significant history of infections (particularly bacterial) of the skin and respiratory tract; also increased susceptibility to periodontal disease
- Mild bleeding tendency associated with platelet dysfunction such as epistaxis, gum/mucosal bleeding, and easy bruising
- Increased risk for hemophagocytic lymphohistiocytosis (HLH) (previously called "CHS accelerated phase"). Clinical findings and diagnostic criteria are the same as those for familial hemophagocytic lymphohistiocytosis.
- Childhood- to early adult-onset neurologic manifestations, including:
- Learning difficulties
- Peripheral neuropathy
- Ataxia
- Parkinsonism
Supportive laboratory findings
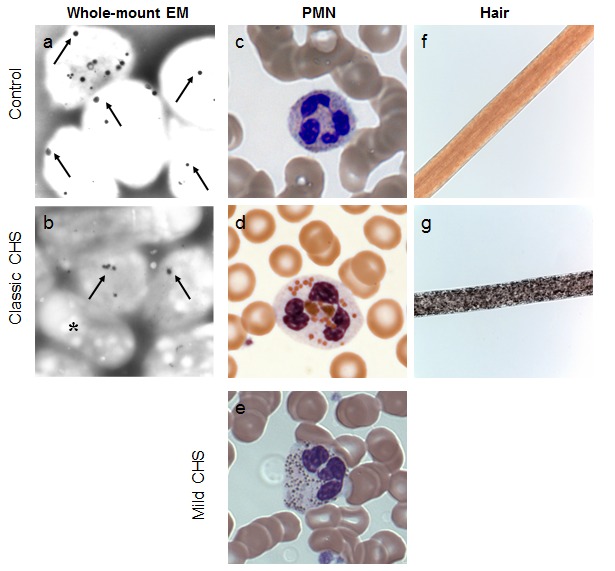
- White blood cell (WBC) giant granules (also called "inclusions"), peroxidase-positive granules primarily in polymorphonuclear neutrophils (PMNs) and to a lesser extent in lymphocytes (Figure 1c-1e), are the most reliable diagnostic criterion for CHS. Nonetheless, giant granules may be overlooked in a routinely evaluated complete blood count (CBC) unless a peripheral smear is reviewed.
- Although the giant granules are seen using routine staining techniques, in some individuals with atypical CHS the presence of these giant granules can be less striking and thus missed by routine evaluation.
- Slide review is optimally conducted by a hematologist or other specialist with experience reviewing blood smears for the presence of abnormal granules.
- Because the finding of WBC giant granules is the most reliable clinical diagnostic criterion for CHS, the combination of any of the other hematologic findings listed below should prompt review of a peripheral blood smear to evaluate for giant granules.
- Other hematologic findings:
- Normal or reduced number of natural killer cells with abnormal (reduced) function
- Neutropenia
- Normal immunoglobulins, complement, antibody production, and delayed hypersensitivity

Other. Pigment clumping on polarized light microscopy hair analysis (Figure 1f, 1g)
Family history is consistent with autosomal recessive inheritance (e.g., affected sibs and/or parental consanguinity). Absence of a known family history does not preclude the diagnosis. Note: When evaluating sibs of a proband for the purpose of the family history, clinicians should have a high level of suspicion for all findings that may be associated with CHS, since presenting manifestations may vary in sibs with the same biallelic LYST pathogenic variants.
Establishing the Diagnosis
The clinical diagnosis of CHS is established in a proband by identification of the pathognomonic giant granules within leukocytes on peripheral blood smear (see Suggestive Findings). The molecular diagnosis of CHS is established in a proband by the identification of biallelic pathogenic (or likely pathogenic) variants in LYST on molecular genetic testing [Sharma et al 2020, Morimoto et al 2023] (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variant" in this GeneReview is understood to include likely pathogenic variants. (2) Identification of biallelic LYST variants of uncertain significance (or of one known LYST pathogenic variant and one LYST variant of uncertain significance) does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing). Gene-targeted testing requires that the clinician determine which gene(s) are likely involved (Option 1), whereas comprehensive genomic testing does not (Option 2).
Option 1
Single-gene testing is an option when laboratory and clinical findings are strongly suggestive of the diagnosis. Sequence analysis of LYST is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
An albinism or neutropenia panel that includes LYST and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible. To date, the majority of LYST pathogenic variants reported (e.g., missense, nonsense) are within the coding region or close to canonical splices and are therefore likely to be identified by exome sequencing.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Chediak-Higashi Syndrome
Clinical Characteristics
Clinical Description
Chediak-Higashi syndrome (CHS) is characterized by partial oculocutaneous albinism (OCA), immunodeficiency, and a mild bleeding tendency. These features are present in nearly all individuals with CHS but to a very variable degree. Affected individuals with severe presentations (i.e., OCA; early-onset, recurrent, severe infections; and a bleeding diathesis) are considered to have "classic" CHS. Individuals with milder phenotypes (e.g., later-onset, milder pigmentary, immunologic, and hematologic features) are considered to have "atypical" CHS (also referred to as "mild" or "adolescent" CHS). Both groups of individuals are at risk of developing hemophagocytic lymphohistiocytosis (HLH), previously called "the accelerated phase," with the highest risk of HLH (~85%) in individuals with classic CHS.
Over time, it has become apparent that the classification of CHS into "classic" vs "atypical" phenotypes is arbitrary, as the considerable overlap of the two groups means that this disorder is best understood as a continuum of severe to milder phenotypes, with the universal feature being the pathognomonic giant granules within leukocytes observed on peripheral blood smear.
Although the proportion of individuals with atypical CHS is unknown [Karim et al 2002, Westbroek et al 2007], it is likely underrecognized. In some individuals with atypical CHS, the neurologic findings may be the predominant manifestation. Additionally, some individuals may not be diagnosed until the third decade of life or later [Weisfeld-Adams et al 2013, Yarnell et al 2020].
Table 2.
Chediak-Higashi Syndrome: Phenotypic Continuum
Partial oculocutaneous albinism (OCA). Pigment dilution, which can involve eyes, hair, and skin, is highly variable.
Reduced iris pigmentation and iris transillumination may be subtle. Affected individuals may have decreased retinal pigmentation and nystagmus. Visual acuity varies from normal to moderately reduced.
The hair has a "silvery" or metallic appearance. Pigment clumping within the shaft of the hair is generally observed by light microscopy (Figure 1g) [Smith et al 2005].
Skin pigment dilution may not be appreciated unless compared to the pigmentation of family members. Individuals with darker skin tone may observe areas with scattered hyper- and hypopigmentation.
Although partial OCA was once thought to be a diagnostic criterion for CHS, at least two individuals with atypical CHS had no evidence of OCA [Introne et al 2017].
Immunodeficiency. Frequent infections usually begin in infancy and are often severe in classic CHS. Individuals with atypical CHS may not have a noticeable increase in severity or frequency of infections.
Bacterial infections are most common, with Staphylococcus and Streptococcus species predominating; viral and fungal infections can also occur [Introne et al 1999]. Infections of the skin and upper respiratory tract are the most common.
Periodontitis, an important manifestation of immunologic dysfunction [Thumbigere Math et al 2018, de Arruda et al 2023], can be the clinical finding that leads to the correct diagnosis [Bailleul-Forestier et al 2008].
Neutropenia may be present and, in some individuals, cycles between normal absolute neutrophil counts and neutropenia (also called "cyclic neutropenia").
Bleeding tendency. The bleeding diathesis in CHS, a result of absent or severely reduced platelet-dense granules, is present in both classic and atypical CHS. Clinical manifestations are generally mild and include epistaxis, gum/mucosal bleeding, and easy bruising. The bleeding diathesis may also be subtle (i.e., generally not requiring medical intervention) and thus may not be identified as a health concern by affected persons. However, with trauma or invasive procedures, bleeding may be more severe and prolonged.
Neurologic involvement. Despite successful hematologic and immunologic outcomes with allogenic hematopoietic stem cell transplantation (HSCT) to treat hematologic findings, neurologic involvement nonetheless manifests by early adulthood.
Neurologic features are similar across the CHS phenotypic spectrum; thus, individuals with classic and atypical CHS cannot be distinguished neurologically [Introne et al 2017]. Due to the wide range of neurologic features that can occur, findings among affected individuals are variable and nonspecific. Likewise, age of onset and disease progression also vary. While learning difficulties may be present in childhood and can be considered developmental in nature, other neurologic signs and symptoms are generally not observed until late adolescence or early adulthood and are progressive. Neurologic findings can include:
- Learning difficulties in childhood [Introne et al 2017, Shirazi et al 2019]
- Sensory neuropathy. Onset in late adolescence or early third decade and slowly progresses to sensorimotor neuropathies and/or diffuse motor neuronopathy [Lehky et al 2017]
- Cerebellar dysfunction. Onset in late adolescence or early adulthood [Introne et al 2017]
- Optic neuropathy. Onset in late adolescence or early adulthood [Desai et al 2016]
- Spastic paraplegia. Onset in early to middle adulthood [Shimazaki et al 2014, Koh et al 2022]
- Tremor, which can include kinetic and postural tremor
- Parkinsonism, including L-dopa-responsive parkinsonism, may occur as early as the second or third decade [Bhambhani et al 2013, Weisfeld-Adams et al 2013]
- Progressive cognitive decline late in the disease course
Hemophagocytic lymphohistiocytosis (HLH; also known as the "accelerated phase") occurs in the majority of individuals with CHS who have not undergone HSCT [Lozano et al 2014] and can occur at any age. Although individuals with atypical CHS are thought to be at lower risk of HLH than individuals with classic CHS, the frequency of occurrence in atypical CHS is unknown.
Originally thought to be a malignancy resembling lymphoma, the "accelerated phase" is now known to be HLH characterized by multiorgan inflammation. Manifestations include fever, lymphadenopathy, hepatosplenomegaly, anemia, neutropenia, and thrombocytopenia.
Triggers of HLH remain unclear. Although infection with Epstein-Barr virus is thought to hasten development of HLH, this relationship has never been proven. Abnormal function of NK cells and cytolytic T cells is also believed to contribute to development of HLH [Jessen et al 2011, Gil-Krzewska et al 2016].
Prognosis. HLH and its complications are the most common cause of mortality in individuals with CHS [Lozano et al 2014].
Genotype-Phenotype Correlations
Clinical phenotypes of CHS have been correlated with classes of LYST pathogenic variants [Karim et al 2002, Zarzour et al 2005, Westbroek et al 2007, Morimoto et al 2023].
- Loss-of-function LYST pathogenic variants (e.g., nonsense, frameshift, canonical splice site, single- or multiexon deletions) are typically associated with classic CHS.
- Missense pathogenic variants and in-frame deletions of LYST are associated with atypical CHS; however, individuals with biallelic missense pathogenic variants with classic CHS and HLH have been reported [Sánchez-Guiu et al 2014].
Prevalence
Fewer than 500 individuals with CHS have been reported [Morimoto et al 2023, Talbert et al 2023].
Exact prevalence is difficult to determine, as some individuals have been reported in the literature more than once. In addition, the broad phenotypic spectrum that has become evident since the early descriptions of CHS suggests that many mildly affected individuals may be underrecognized or underreported.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with germline pathogenic variants in LYST.
Differential Diagnosis
The diagnosis of Chediak-Higashi syndrome (CHS) should be considered in individuals with pigment dilution defects of the hair, skin, or eyes; congenital or transient neutropenia; immunodeficiency; and otherwise unexplained neurologic abnormalities or neurodegeneration. Each of these findings may be variably represented or absent in affected individuals; therefore, heightened suspicion is needed to pursue an accurate diagnosis.
Table 3.
Genes of Interest in the Differential Diagnosis of Chediak-Higashi Syndrome
Management
No clinical practice guidelines for Chediak-Higashi syndrome (CHS) have been published. In the absence of published guidelines, the following recommendations are based on the authors' personal experience managing individuals with this disorder.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Chediak-Higashi syndrome (CHS), the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Chediak-Higashi Syndrome: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
There is no cure for Chediak-Higashi syndrome.
Targeted Therapy
In GeneReviews, a targeted therapy is one that addresses the specific underlying mechanism of disease causation (regardless of whether the therapy is significantly efficacious for one or more manifestation of the genetic condition); would otherwise not be considered without knowledge of the underlying genetic cause of the condition; or could lead to a cure. —ED
Hematopoietic stem cell transplantation (HSCT). The only targeted therapy currently available is HSCT. While HSCT will correct the hematologic and immunologic manifestations of CHS, it does not appear to protect against the development of neurologic manifestations in late adolescence and adulthood.
Preparation for HSCT is often initiated as soon as the diagnosis of CHS is confirmed. HSCT protocols vary by institution.
Best outcomes for HSCT are achieved if initiated prior to the development of hemophagocytic lymphohistiocytosis (HLH); however, if HLH occurs, treatment following the Histiocyte Society HLH-94 protocol is recommended. Once remission of HLH is achieved, HSCT may be performed.
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations in Table 6 are recommended.
Table 6.
Chediak-Higashi Syndrome: Recommended Surveillance
Agents/Circumstances to Avoid
Avoid the following:
- Live vaccines given the risk of infection due to immunodeficiency
- All nonsteroidal anti-inflammatory drugs (NSAIDs) (e.g., aspirin, ibuprofen) given the risk of exacerbating the bleeding tendency
Evaluation of Relatives at Risk
For early diagnosis and treatment. It is appropriate to evaluate the older and younger sibs of a proband as early as possible. Early diagnosis may provide the opportunity to perform HSCT prior to the development of HLH. Evaluations include:
- Molecular genetic testing if the LYST pathogenic variants in the family are known;
- Examination of peripheral blood for the presence of giant granules in white blood cells if the LYST pathogenic variants in the family are not known. (Note: Although the giant granules are seen using routine staining techniques, in some individuals with atypical CHS the presence of these giant granules can be somewhat subtle. A hematologist or a clinician experienced in reviewing blood smears for the presence of these giant granules should review the slide.)
For hematopoietic stem cell donation. Any relative considering stem cell donation should undergo molecular genetic testing to clarify their genetic status so that informed risk vs benefit discussions for both recipient and donor can be incorporated into transplant donor-option decision making. Whenever possible, related donors who do not have a familial LYST pathogenic variant are preferred.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
Based on the limited number of pregnancies in females with CHS reported in the literature to date, pregnancy, labor, and delivery were uneventful [Price et al 1992, Weisfeld-Adams et al 2013]. The infants were healthy.
Bleeding during delivery and the postpartum period are a concern; thus, prior to delivery developing a plan to deal with possible bleeding is recommended.
Therapies Under Investigation
A natural history study at the NIH (Study of Chediak-Higashi Syndrome, NCT00005917) is currently enrolling individuals for longitudinal studies but not offering new therapy.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Chediak-Higashi syndrome (CHS) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected individual are usually heterozygous for a LYST pathogenic variant.
- If a molecular diagnosis has been established in the proband, molecular genetic testing is recommended for the parents of the proband to confirm that both parents are heterozygous for a LYST pathogenic variant and to allow reliable recurrence risk assessment.
- If a pathogenic variant is detected in only one parent and parental identity testing has confirmed biological maternity and paternity, it is possible that one of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Jónsson et al 2017]. If the proband appears to have homozygous pathogenic variants (i.e., the same two pathogenic variants), additional possibilities to consider include:
- A single- or multiexon deletion in the proband that was not detected by sequence analysis and that resulted in the artifactual appearance of homozygosity;
- Uniparental isodisomy for the parental chromosome with the pathogenic variant that resulted in homozygosity for the pathogenic variant in the proband. Two individuals with CHS caused by uniparental disomy of chromosome 1 have been reported [Dufourcq-Lagelouse et al 1999, Manoli et al 2010].
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for a LYST pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Sibs who inherit the same biallelic LYST pathogenic variants as the proband may present with discrepant phenotypes [Morimoto et al 2023].
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. The offspring of an individual with CHS are obligate heterozygotes (carriers) for a LYST pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a LYST pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the LYST pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Once the LYST pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- Hermansky-Pudlak Syndrome Network, Inc.Phone: 800-789-9HPSFax: 516-624-0640Email: info@hpsnetwork.org
- International Patient Organization for Primary Immunodeficiencies (IPOPI)United KingdomPhone: +44 01503 250 668Fax: +44 01503 250 668Email: info@ipopi.org
- National Organization for Albinism and Hypopigmentation (NOAH)Phone: 800-473-2310 (US and Canada); 603-887-2310Fax: 603-887-6049Email: info@albinism.org
- European Society for Immunodeficiencies (ESID) RegistryEmail: esid-registry@uniklinik-freiburg.de
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Chediak-Higashi Syndrome: Genes and Databases
Table B.
OMIM Entries for Chediak-Higashi Syndrome (View All in OMIM)
Molecular Pathogenesis
LYST encodes the cytoplasmic protein lysosomal-trafficking regulator (LYST), also called CHS1. It has been hypothesized that LYST has a role in the regulation of membrane fusion events and lysosomal size [Tanabe et al 2000, Tchernev et al 2002, Möhlig et al 2007, Morimoto et al 2007]. Studies in Drosophila have suggested that LYST plays a role in regulating lysosome-related organelle (LRO) formation and in facilitating centrosome maturation and positioning [Lattao et al 2021]. Another study suggested LYST involvement in autophagosome lysosome reformation facilitates fission of autolysosome tubules for lysosomal homeostasis [Serra-Vinardell et al 2023]. However, the precise biologic role of LYST remains unknown.
Mechanism of disease causation. Loss of function
Variants of uncertain significance (VUS). As the use of broader genomic sequencing increases, identification of VUS is also rising. For individuals identified to have VUS in LYST, it is important to correlate the genetic findings with the clinical features following a comprehensive evaluation, specifically taking into consideration the finding of giant granules within leukocytes on the peripheral blood smear (see Suggestive Findings). If a blood smear is unavailable, light microscopy of the hair showing the characteristic pigment clumping throughout the hair shaft may aid in interpretation of VUS.
Chapter Notes
Author Notes
Dr Toro is a movement disorder neurologist who works with the National Institutes of Health Undiagnosed Diseases Program.
Dr Morimoto is a scientist who performs translational research in the National Institutes of Health Undiagnosed Diseases Program.
Dr Malicdan is a neurologist and a scientist whose main interest is the study of rare diseases. She performs basic and translational research on rare diseases in the National Institutes of Health Undiagnosed Diseases Program.
Dr Adams is a pediatrician, medical geneticist, and biochemical geneticist who performs clinical and basic research on rare diseases at the National Institutes of Health.
Dr Introne (vog.hin@enortniw) is a pediatrician and medical and biochemical geneticist who performs clinical research on rare diseases at the National Institutes of Health. Dr Introne is actively involved in clinical research regarding Chediak-Higashi syndrome and Hermansky-Pudlak syndrome. She would be happy to communicate with persons who have questions regarding the diagnosis of these disorders or other considerations.
Acknowledgments
The authors would like to gratefully acknowledge the patients with Chediak-Higashi syndrome and their families for contributing to our understanding of CHS over the years. Special thanks to the Chediak-Higashi Syndrome Association and the Hermansky-Pudlak Syndrome Network for their long-standing dedication to research and patient advocacy.
The authors would like to respectfully acknowledge the authors of the Familial Hemophagocytic Lymphohistiocytosis GeneReview for allowing us to link to their GeneReview and providing expert information on the clinical presentation, diagnosis, and management of HLH.
This work was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, Bethesda, Maryland.
Author History
David R Adams, MD, PhD (2009-present)
Gretchen A Golas, RN, MS, CRNP; National Human Genome Research Institute (2009-2018)
Wendy J Introne, MD (2009-present)
May Christine Malicdan, MD, PhD (2018-present)
Marie Morimoto, PhD (2023-present)
Elena-Raluca Nicoli, PharmR, PhD; National Human Genome Research Institute (2018-2023)
Camilo Toro, MD (2018-present)
Wendy Westbroek, PhD; National Human Genome Research Institute (2009-2018)
Revision History
- 21 December 2023 (bp) Comprehensive update posted live
- 5 July 2018 (ma) Comprehensive update posted live
- 15 January 2015 (me) Comprehensive update posted live
- 16 February 2012 (me) Comprehensive update posted live
- 3 March 2009 (me) Review posted live
- 2 October 2008 (wji) Original submission
Note: Pursuant to 17 USC Section 105 of the United States Copyright Act, the GeneReview "Chediak-Higashi Syndrome" is in the public domain in the United States of America.
References
Literature Cited
- Bailleul-Forestier I, Monod-Broca J, Benkerrou M, Mora F, Picard B. Generalized periodontitis associated with Chediak-Higashi syndrome. J Periodontol. 2008;79:1263–70. [PubMed: 18597610]
- Bergsten E, Horne A, Aricó M, Astigarraga I, Egeler RM, Filipovich AH, Ishii E, Janka G, Ladisch S, Lehmberg K, McClain KL, Minkov M, Montgomery S, Nanduri V, Rosso D, Henter JI. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017;130:2728–38. [PMC free article: PMC5785801] [PubMed: 28935695]
- Bhambhani V, Introne WJ, Lungu C, Cullinane A, Toro C. Chediak-Higashi syndrome presenting as young-onset levodopa-responsive parkinsonism. Mov Disord. 2013;28:127–9. [PMC free article: PMC3581862] [PubMed: 23436631]
- Certain S, Barrat F, Pastural E, Le Deist F, Goyo-Rivas J, Jabado N, Benkerrou M, Seger R, Vilmer E, Beullier G, Schwarz K, Fischer A, de Saint Basile G. Protein truncation test of LYST reveals heterogenous mutations in patients with Chediak-Higashi syndrome. Blood. 2000;95:979-83. [PubMed: 10648412]
- de Arruda JAA, Sousa-Neto SS, Abreu LG, Schuch LF, Souza VG, Alves TVL, Martins-Andrade B, Shetty SS, Monteiro JLGC, Mendonça EF, Mesquita RA, Callou G. Oral manifestations of Chediak-Higashi syndrome: A systematic review. Dis Mon. 2023;69:101356. [PubMed: 35414415]
- Desai N, Weisfeld-Adams JD, Brodie SE, Cho C, Curcio CA, Lublin F, Rucker JC. Optic neuropathy in late-onset neurodegenerative Chédiak-Higashi syndrome. Br J Ophthalmol. 2016;100:704-7. [PubMed: 26307451]
- Dufourcq-Lagelouse R, Lambert N, Duval M, Viot G, Vilmer E, Fischer A, Prieur M, de Saint Basile G. Chediak-Higashi syndrome associated with maternal uniparental isodisomy of chromosome 1. Eur J Hum Genet. 1999;7:633–7. [PubMed: 10482950]
- Eapen M, DeLaat CA, Baker KS, Cairo MS, Cowan MJ, Kurtzberg J, Steward CG, Veys PA, Filipovich AH. Hematopoietic cell transplantation for Chediak-Higashi syndrome. Bone Marrow Transplantation. 2007;39:411–5. [PubMed: 17293882]
- Filipovich AH, Chandrakasan S. Pathogenesis of hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 2015;29:895–902. [PubMed: 26461149]
- Fusaro M, Rosain J, Grandin V, Lambert N, Hanein S, Fourrage C, Renaud N, Gil M, Chevalier S, Chahla WA, Bader-Meunier B, Barlogis V, Blanche S, Boutboul D, Castelle M, Comont T, Diana JS, Fieschi C, Galicier L, Hermine O, Lefèvre-Utile A, Malphettes M, Merlin E, Oksenhendler E, Pasquet M, Suarez F, André I, Béziat V, De Saint Basile G, De Villartay JP, Kracker S, Lagresle-Peyrou C, Latour S, Rieux-Laucat F, Mahlaoui N, Bole C, Nitschke P, Hulier-Ammar E, Fischer A, Moshous D, Neven B, Alcais A, Vogt G, Bustamante J, Picard C. Improving the diagnostic efficiency of primary immunodeficiencies with targeted next-generation sequencing. J Allergy Clin Immunol. 2021;147:734-37. [PubMed: 32531373]
- Gil-Krzewska A, Wood SM, Murakami Y, Nguyen V, Chiang SCC, Cullinane AR, Peruzzi G, Gahl WA, Coligan JE, Introne WJ, Bryceson YT, Krzewski K. Chediak-Higashi syndrome: lysosomal trafficking regulator domains regulate exocytosis of lytic granules but not cytokine secretion by natural killer cells. J Allergy Clin Immunol. 2016;137:1165–77. [PMC free article: PMC4826811] [PubMed: 26478006]
- Hartz B, Marsh R, Rao K, Henter JI, Jordan M, Filipovich L, Bader P, Beier R, Burkhardt B, Meisel R, Schulz A, Winkler B, Albert MH, Greil J, Karasu G, Woessmann W, Corbacioglu S, Gruhn B, Holter W, Kühl JS, Lang P, Seidel MG, Veys P, Löfstedt A, Ammann S, Ehl S, Janka G, Müller I, Lehmberg K. The minimum required level of donor chimerism in hereditary hemophagocytic lymphohistiocytosis. Blood. 2016;127:3281–90. [PMC free article: PMC5291300] [PubMed: 27099148]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab. 1999;68:283–303. [PubMed: 10527680]
- Introne WJ, Westbroek W, Groden CA, Bhambhani V, Golas GA, Baker EH, Lehky TJ, Snow J, Ziegler SG, Malicdan MC, Adams DR, Horward HM, Hess RA, Huizing M, Gahl WA, Toro C. Neurologic involvement in patients with atypical Chediak-Higashi disease. Neurology. 2017;88:e57–e65. [PMC free article: PMC5584077] [PubMed: 28193763]
- Jessen B, Maul-Pavicic A, Ufheil H, Vraetz T, Enders A, Lehmberg K, Langler A, Gross-Wieltsch U, Bay A, Kaya Z, Bryceson YT, Koscielniak E, Badawy S, Davies G, Hufnagel M, Schmitt-Graeff A, Aichele P, Zur Stadt U, Schwarz K, Ehl S. Subtle differences in CTL cytotoxicity determine susceptibility to hemophagocytic lymphohistiocytosis in mice and humans with Chediak-Higashi syndrome. Blood. 2011;118:4620–9. [PubMed: 21878672]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519-22. [PubMed: 28959963]
- Karim MA, Suzuki K, Fukai K, Oh J, Nagle DL, Moore KJ, Barbosa E, Falik-Borenstein T, Filipovich A, Ishida Y, Kivrikko S, Klein C, Kreuz F, Levin A, Miyajima H, Regueiro J, Russo C, Uyama E, Vierimaa O, Spritz RA. Apparent genotype-phenotype correlation in childhood, adolescent, and adult Chediak Higashi syndrome. Am J Med Genet. 2002;108:16–22. [PubMed: 11857544]
- Koh K, Tsuchiya M, Ishiura H, Shimazaki H, Nakamura T, Hara H, Suzuyama K, Takahashi M, Tsuji S, Takiyama Y; Japan Spastic Paraplegia Research Consortium. Chédiak-Higashi syndrome presenting as a hereditary spastic paraplegia. J Hum Genet. 2022;67:119-21. [PubMed: 34483340]
- Kuptanon C, Morimoto M, Nicoli ER, Stephen J, Yarnell DS, Dorward H, Owen W, Parikh S, Ozbek NY, Malbora B, Ciccone C, Gunay-Aygun M, Gahl WA, Introne WJ, Malicdan MCV. cDNA sequencing increases the molecular diagnostic yield in Chediak-Higashi syndrome. Front Genet. 2023;14:1072784. [PMC free article: PMC10031035] [PubMed: 36968585]
- Lattao R, Rangone H, Llamazares S, Glover DM. Mauve/LYST limits fusion of lysosome-related organelles and promotes centrosomal recruitment of microtubule nucleating proteins. Dev Cell. 2021;56:1000-1013.e6. [PMC free article: PMC8024676] [PubMed: 33725482]
- Lehky TJ, Groden C, Lear B, Toro C, Introne WJ. Peripheral nervous system manifestations of Chediak-Higashi disease. Muscle Nerve. 2017;55:359–65. [PMC free article: PMC5243934] [PubMed: 27429304]
- Lozano ML, Rivera J, Sánchez-Guiu I, Vicente V. Towards the targeted management of Chediak-Higashi syndrome. Orphanet J Rare Dis. 2014;9:132. [PMC free article: PMC4243965] [PubMed: 25129365]
- Manoli I, Golas G, Westbroek W, Vilboux T, Markello TC, Introne W, Maynard D, Pederson B, Tsilou E, Jordan MB, Hart PS, White JG, Gahl WA, Huizing M. Chediak-Higashi syndrome with early developmental delay resulting from paternal heterodisomy of chromosome 1. Am J Med Genet A. 2010;152A:1474–83. [PMC free article: PMC2947940] [PubMed: 20503323]
- Marsh RA, Vaughn G, Kim MO, Li D, Jodele S, Joshi S, Mehta PA, Davies SM, Jordan MB, Bleesing JJ, Filipovich AH. Reduced-intensity conditioning significantly improves survival of patients with hemophagocytic hymphohistiocytosis undergoing allogenic hematopoietic cell transplantation. Blood. 2010;116:5824–31. [PubMed: 20855862]
- McCusker C, Warrington R. Primary immunodeficiency. Allergy Asthma Clin Immunol. 2011;7:S11. [PMC free article: PMC3245434] [PubMed: 22165913]
- Möhlig H, Mathieu S, Thon L, Frederiksen MC, Ward DM, Kaplan J, Schütze S, Kabelitz D, Adam D. The WD repeat protein FAN regulates lysosome size independent from abnormal downregulation/membrane recruitment of protein kinase C. Exp Cell Res. 2007;313:2703–18. [PMC free article: PMC2988431] [PubMed: 17512928]
- Morimoto M, Nicoli ER, Kuptanon C, Roney JC, Serra-Vinardell J, Sharma P, Adams DR, Gallin JI, Holland SM, Rosenzweig SD, Barbot J, Ciccone C, Huizing M, Toro C, Gahl WA, Introne WJ, Malicdan MCV. Spectrum of LYST mutations in Chediak-Higashi syndrome: a report of novel variants and a comprehensive review of the literature. J Med Genet. 2023. Epub ahead of print. [PubMed: 37788905]
- Morimoto M, Tanabe F, Kasai H, Ito M. Effect of a thiol proteinase inhibitor, E-64-d, on susceptibility to infection with Staphylococcus aureus in Chediak-Higashi syndrome (beige) mice. Int Immunopharmacol. 2007;7:973–80. [PubMed: 17499200]
- Price FV, Legro RS, Watt-Morse M, Kaplan SS. Chediak-Higashi syndrome in pregnancy. Obstet Gynecol. 1992;79:804–6. [PubMed: 1565370]
- Principi N, Esposito S. Vaccine use in primary immunodeficiency disorders. Vaccine. 2014;32:3725–31. [PubMed: 24837766]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Sánchez-Guiu I, Antón AI, García-Barberá N, Navarro-Fernández J, Martínez C, Fuster JL, Couselo JM, Ortuño FJ, Vicente V, Rivera J, Lozano ML. Chediak-Higashi syndrome: description of two novel homozygous missense mutations causing divergent clinical phenotype. Eur J Haematol. 2014;92:49–58. [PubMed: 24112114]
- Serra-Vinardell J, Sandler MB, De Pace R, Manzella-Lapeira J, Cougnoux A, Keyvanfar K, Introne WJ, Brzostowski JA, Ward ME, Gahl WA, Sharma P, Malicdan MCV. LYST deficiency impairs autophagic lysosome reformation in neurons and alters lysosome number and size. Cell Mol Life Sci. 2023;80:53. [PMC free article: PMC11072721] [PubMed: 36707427]
- Sharma P, Nicoli ER, Serra-Vinardell J, Morimoto M, Toro C, Malicdan MCV, Introne WJ. Chediak-Higashi syndrome: a review of the past, present, and future. Drug Discov Today Dis Models. 2020;31:31-6. [PMC free article: PMC7793027] [PubMed: 33424983]
- Shimazaki H, Honda J, Naoi T, Namekawa M, Nakano I, Yazaki M, Nakamura K, Yoshida K, Ikeda S, Ishiura H, Fukuda Y, Takahashi Y, Goto J, Tsuji S, Takiyama Y. Autosomal-recessive complicated spastic paraplegia with a novel lysosomal trafficking regulator gene mutation. J Neurol Neurosurg Psychiatry. 2014;85:1024–8. [PubMed: 24521565]
- Shirazi TN, Snow J, Ham L, Raglan GB, Wiggs EA, Summers AC, Toro C, Introne WJ. The neuropsychological phenotype of Chediak-Higashi disease. Orphanet J Rare Dis. 2019;14:101. [PMC free article: PMC6503440] [PubMed: 31060595]
- Smith VV, Anderson G, Malone M, Sebire NJ. Light microscopic examination of scalp hair samples as an aid in the diagnosis of paediatric disorders: retrospective review of more than 300 cases from a single centre. J Clin Pathol. 2005;58:1294–8. [PMC free article: PMC1770794] [PubMed: 16311350]
- Sobh A, Bonilla FA. Vaccination in primary immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016;4:1066–75. [PubMed: 27836056]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Talbert ML, Malicdan MCV, Introne WJ. Chediak-Higashi syndrome. Curr Opin Hematol. 2023;30:144-51. [PMC free article: PMC10501739] [PubMed: 37254856]
- Tanabe F, Cui SH, Ito M. Abnormal down-regulation of PKC is responsible for giant granule formation in fibroblasts from CHS (beige) mice--a thiol proteinase inhibitor, E-64-d, prevents giant granule formation in beige fibroblasts. J Leukoc Biol. 2000;67:749–55. [PubMed: 10811017]
- Tchernev VT, Mansfield TA, Giot L, Kumar AM, Nandabalan K, Li Y, Mishra VS, Detter JC, Rothberg JM, Wallace MR, Southwick FS, Kingsmore SF. The Chediak-Higashi protein interacts with SNARE complex and signal transduction proteins. Mol Med. 2002;8:56–64. [PMC free article: PMC2039936] [PubMed: 11984006]
- Thumbigere Math V, Rebouças P, Giovani PA, Puppin-Rontani RM, Casarin R, Martins L, Wang L, Krzewski K, Introne WJ, Somerman MJ, Nociti FH Jr, Kantovitz KR. Periodontitis in Chédiak-Higashi syndrome: an altered immunoinflammatory response. JDR Clin Trans Res. 2018;3:35-46. [PMC free article: PMC5734460] [PubMed: 29276776]
- Trottestam H, Beutel K, Meeths M, Carlsen N, Heilmann C, Pasić S, Webb D, Hasle H, Henter JI. Treatment of the X-linked lymphoproliferative, Griscelli and Chediak-Higashi syndromes by HLH directed therapy. Pediatr Blood Cancer. 2009;52:268–72. [PubMed: 18937330]
- Weisfeld-Adams JD, Mehta L, Rucker JC, Dembitzer FR, Szporn A, Lublin FD, Introne WJ, Bhambhani V, Chicka MC, Cho C. Atypical Chediak-Higashi syndrome with attenuated phenotype: three adult siblings homozygous for a novel LYST deletion and with neurodegenerative disease. Orphanet J Rare Dis. 2013;8:46. [PMC free article: PMC3610301] [PubMed: 23521865]
- Westbroek W, Adams D, Huizing M, Koshoffer A, Dorward H, Tinloy B, Parkes J, Helip-Wooley A, Kleta R, Tsilou E, Duvernay P, Digre KB, Creel DJ, White JG, Boissy RE, Gahl WA. The severity of cellular defects in Chediak Higashi syndrome correlate with the molecular genotype and clinical phenotype. J Invest Dermatol. 2007;127:2674–7. [PubMed: 17554367]
- Yarnell DS, Roney JC, Teixeira C, Freitas MI, Cipriano A, Leuschner P, Krzewski K, Stephen J, Dorward H, Gahl WA, Gochuico BR, Toro C, Malicdan MC, Introne WJ. Diagnosis of Chediak Higashi disease in a 67-year old woman. Am J Med Genet A. 2020;182:3007-13. [PubMed: 32990340]
- Zarzour W, Kleta R, Frangoul H, Suwannarat P, Jeong A, Kim SY, Wayne AS, Gunay-Aygun M, White J, Filipovich AH, Gahl WA. Two novel CHS1 (LYST) mutations: clinical correlations in an infant with Chediak-Higashi syndrome. Mol Genet Metab. 2005;85:125–32. [PubMed: 15896657]
Publication Details
Author Information and Affiliations
National Institutes of Health
Bethesda, Maryland
National Institutes of Health
Bethesda, Maryland
National Institutes of Health
Bethesda, Maryland
National Institutes of Health
Bethesda, Maryland
National Institutes of Health
Bethesda, Maryland
Publication History
Initial Posting: March 3, 2009; Last Update: December 21, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Toro C, Morimoto M, Malicdan MC, et al. Chediak-Higashi Syndrome. 2009 Mar 3 [Updated 2023 Dec 21]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.