Summary
Clinical characteristics.
The NSDHL-related disorders include: CHILD (congenital hemidysplasia with ichthyosiform nevus and limb defects) syndrome, an X-linked condition that is usually male lethal during gestation and thus predominantly affects females; and CK syndrome, an X-linked disorder that affects males.
- CHILD syndrome is characterized by unilateral distribution of ichthyosiform (yellow scaly) skin lesions and ipsilateral limb defects that range from shortening of the metacarpals and phalanges to absence of the entire limb. Intellect is usually normal. The ichthyosiform skin lesions are usually present at birth or in the first weeks of life; new lesions can develop in later life. Nail changes are also common. The heart, lung, and kidneys can also be involved.
- CK syndrome (named for the initials of the original proband) is characterized by mild to severe cognitive impairment and behavior problems (aggression, attention deficit hyperactivity disorder, and irritability). All affected males reported have developed seizures in infancy and have cerebral cortical malformations and microcephaly. All have distinctive facial features, a thin habitus, and relatively long, thin fingers and toes. Some have scoliosis and kyphosis. Strabismus is common. Optic atrophy is also reported.
Diagnosis/testing.
The diagnosis of CHILD syndrome is established in a proband by identification of an NSDHL pathogenic variant that results in loss of functional NSDHL protein. The diagnosis of CK syndrome is established in a proband by identification of a "hypomorphic" NSDHL pathogenic variant that results in partial loss of functional NSDHL protein.
Management.
Treatment of manifestations:
- CHILD syndrome. No one therapy described to date appears to ameliorate the cutaneous findings for every reported individual with CHILD syndrome. Lactic acid 12% skin creams or lotions can reduce itching, and urea skin creams can reduce dryness. Treatment of an inflammatory nevus by grafting skin obtained from a contralateral unaffected region has been successful. Oral aromatic retinoids (etretinate) used to ameliorate cutaneous symptoms have been found to be of limited use and not well tolerated. Topical statins may be beneficial for the treatment of inflammatory nevus. Scoliosis and joint contractures are treated with braces and/or corrective surgery.
- CK syndrome. Behavior modification and/or drug therapy to control aggression and help with ADHD symptoms; anti-seizure medication to control seizures.
Surveillance:
- CHILD syndrome. Monitoring for new cutaneous lesions and musculoskeletal deformities such as scoliosis and joint contractures.
- CK syndrome. Monitoring for the effectiveness of AEDs in controlling seizures and for the development of scoliosis/kyphosis.
Genetic counseling.
The NSDHL-related disorders are inherited in an X-linked manner. No affected male has reproduced.
- CHILD syndrome is usually male lethal during gestation. Affected females have a 50% chance of transmitting the NSDHL pathogenic variant in each pregnancy; however, the expected live-born distribution of persons at risk for CHILD syndrome is 33% unaffected females, 33% affected females, and 33% unaffected males.
- CK syndrome is diagnosed in males. Heterozygous females have a 50% chance of transmitting the NSDHL pathogenic variant in each pregnancy; males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will have normal physical features, intellect, and brain imaging but may display behavioral problems such as irritability and aggression.
Testing of at-risk female relatives and prenatal testing for pregnancies at increased risk for an NSDHL-related disorder are possible if the pathogenic variant has been identified in the family.
GeneReview Scope
Table
CHILD syndrome CK syndrome
Diagnosis
Suggestive Findings
An NSDHL-related disorder should be suspected in an individual with features of CHILD (congenital hemidysplasia with ichthyosiform nevus and limb defects) syndrome (typically in females) and CK syndrome (intellectual disability and associated features in males; CK = initials of the original proband) as follows.
CHILD syndrome
- Unilateral distribution of ichthyosiform nevus
- Limb defects ipsilateral to the skin lesions
- Punctate calcifications of cartilaginous structures
- Visceral malformations
- Central nervous system anomalies
CK syndrome [du Souich et al 2009, McLarren et al 2010, Preiksaitiene et al 2015]
- Central nervous system (CNS) findings: mild to severe intellectual disability, microcephaly, cerebral cortical malformations, spasticity, and seizures
- Characteristic craniofacial features: almond-shaped and upslanted palpebral fissures, prominent nasal bridge, high arched palate, crowded dentition, micrognathia, and plagiocephaly
- Asthenic habitus
Establishing the Diagnosis
Male proband. The diagnosis of an NSDHL-related disorder is established in a male proband with the identification of a hemizygous pathogenic variant in NSDHL by molecular genetic testing (see Table 1).
Female proband. The diagnosis of an NSDHL-related disorder is usually established in a female proband with the identification of a heterozygous pathogenic variant in NSDHL by molecular genetic testing (see Table 1).
Note: Animal models show that male conceptuses with a severe NSDHL loss-of-function allele die early in gestation, explaining the fact that with few exceptions individuals with CHILD syndrome are female [Bornholdt et al 2005, Bittar et al 2006].
Identification of a "hypomorphic" NSDHL pathogenic variant that results in partial loss of functional NSDHL protein confirms the diagnosis of CK syndrome [McLarren et al 2010].
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing, exome array) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Because the phenotype of NSDHL-related disorders is broad, individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of NSDHL-related disorders has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and laboratory findings suggest the diagnosis of NSDHL-related disorders molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of NSDHL detects small intragenic deletions/insertions and missense, nonsense, and splice site variants. If no pathogenic variant is found perform gene-targeted deletion/duplication analysis to detect intragenic deletions or duplications.
- A multigene panel that includes NSDHL and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see Table 1).
Option 2
When the diagnosis of NSDHL-related disorders is not considered because an individual has atypical phenotypic features, comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible. Exome array (when clinically available) may be considered if exome sequencing is not diagnostic.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in NSDHL-Related Disorders
Clinical Characteristics
Clinical Description
CHILD Syndrome
CHILD syndrome (Figure 1) is characterized by unilateral distribution of ichthyosiform skin lesions and ipsilateral limb defects (see Bornholdt et al [2005] for a summary of features). The skin and skeletal involvement can be right-sided (seen in ~2/3 of individuals), left-sided, or bilateral [König et al 2002, Hummel et al 2003, Mi et al 2015]. Based on mouse studies and family observation, CHILD-associated NSDHL pathogenic variants are usually lethal to males during gestation.
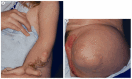
Figure 1.
Photographs of a female with CHILD syndrome A. Upper left limb. Note the forearm hypoplasia, ectrodactyly, onychodystrophy, and characteristic ichthyosiform skin lesions with yellow scales.
Early death of affected females is usually the result of cardiovascular malformations.
A few males with CHILD syndrome have been reported [Zellweger & Uehlinger 1948, Happle et al 1996]. The male reported by Happle et al [1996] had the typical skin findings seen in females with CHILD syndrome and was developmentally normal. He was mosaic for the c.262C>T (p.Arg88Ter) pathogenic variant in NSDHL [Bornholdt et al 2005].
Dermatologic findings
- Ichthyosiform nevus. The hallmark of CHILD syndrome is the presence of ichthyosiform skin lesions with yellow scales and a sharp demarcation in the midline of the body. The initial ichthyosiform skin lesions are evident at birth or in the first weeks of life; new lesions may develop in later life [Happle et al 1980]. The face is usually spared; scalp alopecia has been reported [Hummel et al 2003]. Most skin lesions improve spontaneously, but some can cause lifelong morbidity. Other skin lesions can develop after infancy at sites of injury such as a surgical wound.Histologically the skin lesions exhibit hyperkeratosis, parakeratosis, and acanthosis as well as inflammatory and lipid-laden infiltrates within the dermal papillae [Hebert et al 1987, Hashimoto et al 1995]. The skin lesions from persons with NSDHL pathogenic variants can be distinguished histologically and biochemically from those with chondrodysplasia punctata 2, X-linked, in which unilateral skin and skeletal lesions can occur.Occasionally, heterozygous females present with comparatively minor skin lesions such as Blaschko-linear inflammatory scaly lesions, patchy alopecia, or nail changes. Regardless, the specific finding of an ichthyosiform nevus should always raise the possibility of heterozygosity for an NSDHL pathogenic variant. In some females, an ichthyosiform nevus can be present without any associated symptoms of CHILD syndrome in a woman at risk of having a daughter with typical CHILD syndrome [Happle et al 1995]. The relative severity of disease in studied organs reflects the skewing of X-chromosome inactivation [König, unpublished results].
- Verruciform xanthoma-like lesions. Although rare, these types of lesions were reported in a girl age nine years with CHILD syndrome harboring a large deletion of NSDHL exons 3 and 4 [Yu et al 2018].
- Nails. Onychodystrophy and periungual hyperkeratosis are common.
Skeletal features
- Limbs. Ipsilateral hypoplasia of the limbs varies from shortening of metacarpals and phalanges to absence of the entire limb [Happle et al 1980]. Incomplete development or absence of vertebrae, ribs, and long bones has also been reported [Bornholdt et al 2005].
- Other skeletal defects (generally evident in infancy) include scoliosis and joint contractures.
- Punctate calcifications of cartilaginous structures. Unilateral punctate epiphyseal calcifications in the pelvis, ribs, vertebrae, and extremities have been reported [Happle et al 1980] and are usually seen in the affected limb or body part [Hashimoto et al 1995, Hummel et al 2003]. These can be visible on x-ray examination in infancy. In one child, the punctate calcifications were reported to have disappeared completely by age two years [Happle et al 1980]; however, it is not known whether this is the case for every affected child. Ipsilateral stippling has also been observed in the sella turcica and the laryngeal, nasal, and thyroid cartilage [Happle et al 1980, Grange et al 2000].
Other structural anomalies
- CNS anomalies include unilateral hypoplasia or underdevelopment of the brain, lissencephaly type II, and cerebellar malformation [Tang & McCreadie 1974, Schmidt-Sidor et al 2008]. Hypoplasia of cranial nerves V, VII, VIII, IX, and X and the spinal cord was identified on autopsy in the same individual reported by Tang & McCreadie [1974]. The individual reported by Schmidt-Sidor et al [2008] showed multiple left-sided brain anomalies as a consequence of disturbances in proliferation and migration. The Virchow-Robin spaces of the left parietal lobe were locally enlarged in an affected female reported by Yu et al [2018].Intellect is usually normal; some reported individuals have intellectual disability [Baden & Rex 1970].
- Heart defects include septal defects [König et al 2002], unilateral ventricle [Falek et al 1968], and a single coronary ostium [Tang & McCreadie 1974].
- Lung hypoplasia, observed in several individuals [Tang & McCreadie 1974, Bornholdt et al 2005], can cause respiratory compromise and death [Hummel et al 2003].
- Renal findings range from unilateral hydronephrosis to renal agenesis. The frequency of these is unknown.
Other findings. Reported additional findings include hearing loss, absence of facial muscles, and unilateral hypoplasia of the thyroid gland, adrenal glands, ovaries, and fallopian tubes [Happle et al 1980, König et al 2002]. Bilateral optic atrophy has been reported in one individual [Knape et al 2010], as have thrombocytosis and congenital bilateral dislocation of the hip [Chander et al 2010]. Small intestinal mucosal xanthoma was reported in an individual with CHILD syndrome [Ryan et al 2013].
CK Syndrome
CK syndrome (Figure 2) is an X-linked intellectual disability syndrome that affects males. Although 24 affected males from three unrelated families have been identified and fully evaluated, characterization of the syndrome remains limited.
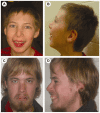
Figure 2.
A male age 11 years (A, B) and a male age 22 years (C,D) with CK syndrome. Note the long thin face, epicanthal folds, almond-shaped palpebral fissures, prominent nasal bridge, and micrognathia. The long thin face becomes more apparent with age.
Development. Affected males have mild to severe intellectual disability. Most cannot speak.
Behavior. Most manifest aggression, attention deficit hyperactivity disorder (ADHD), and irritability. These behaviors appear in infancy and early childhood. According to the Autism Diagnostic Review (ADI-R) and the Autism Diagnostic Observation Schedule (ADOS), affected males do not fulfill the criteria for an autism spectrum disorder.
Neurologic findings. All affected males have developed seizures in infancy. These range from multiple daily episodes of brief unresponsiveness associated with staring and facial and/or limb twitching to prolonged generalized tonic-clonic seizures. These likely arise from cerebral cortical malformations which, by MRI examination, are most consistent with polymicrogyria (see Polymicrogyria Overview). Spasticity, tetraparesis, and development of contractures have also been reported.
Craniofacial. Affected males have a long thin face, plagiocephaly, almond-shaped and upslanted palpebral fissures, prominent nasal bridge, high palate, dental crowding, and micrognathia. The ears are normally shaped but rotated posteriorly.
Growth and skeletal. All affected males have microcephaly (<3 SD to <2 SD), a thin habitus, and relatively long, thin fingers and toes. Some have scoliosis and kyphosis. The height of affected individuals is average for parental heights.
Ocular findings. Strabismus is common. Optic atrophy is also seen.
Analyte testing associated with CHILD syndrome and CK syndrome
- When cultured in cholesterol-depleted medium, lymphoblastoid cells of individuals with CHILD syndrome and CK syndrome have increased levels of methyl- and carboxy-sterols and slightly decreased levels of cholesterol [Grange et al 2000, Hummel et al 2003, McLarren et al 2010].
- In individuals with CHILD syndrome, sterol analysis of skin flakes collected from an affected area show elevated levels of methyl- and carboxy-sterols [RI Kelley, personal communication].
- Serum concentrations of methyl-sterol and cholesterol are almost always normal in individuals with CHILD syndrome and CK syndrome.
Heterozygous females. Females heterozygous for an NSDHL pathogenic variant have normal physical features, intellect, and brain imaging but display behavioral problems including irritability and aggression [Herman & Kratz 2012]. Since heterozygous females have normal plasma cholesterol and plasma 24S-hydroxycholesterol levels, du Souich et al [2012] hypothesized that methyl-sterol accumulation accounts for the behavioral and cognitive problems.
Genotype-Phenotype Correlations
CHILD syndrome. Phenotypic variability within the spectrum of CHILD syndrome does not strictly correlate with the predicted severity of NSDHL pathogenic variants [Bornholdt et al 2005, Mi et al 2015].
CK syndrome. The three reported pathogenic variants (c.696_698del, c.1098dup, and c.455G>A) are associated with the same phenotype in affected males.
Penetrance
Incomplete penetrance has not been reported for CHILD syndrome; therefore, the penetrance is probably very high. Of note, expressivity is highly variable; in affected females, CHILD syndrome may manifest as minor skin changes only.
Penetrance is probably 100% in males with CK syndrome.
Nomenclature
CHILD is an acronym for congenital hemidysplasia with ichthyosiform nevus and limb defects. CHILD syndrome was first reported in 1903 by Dr Otto Sachs [Bittar & Happle 2004].
CK syndrome represents the initials of the original proband. CK syndrome was first described by du Souich et al [2009].
Prevalence
The prevalence of CHILD syndrome is unknown; more than 60 individuals have been reported thus far.
The prevalence of CK syndrome is unknown; it is thought to be rare.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with pathogenic variants in NSDHL.
Differential Diagnosis
See Table 2 (CHILD syndrome) and Table 3 (CK syndrome).
Table 2.
Disorders to Consider in the Differential Diagnosis of CHILD Syndrome
Table 3.
Disorders to Consider in the Differential Diagnosis of CK Syndrome
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with CHILD syndrome, the evaluations summarized in Table 4 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with CHILD Syndrome
To establish the extent of disease and needs in an individual diagnosed with CK syndrome, the evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
Recommended Evaluations Following Initial Diagnosis in Individuals with CK Syndrome
Treatment of Manifestations
CHILD syndrome
- Topical treatments including lactic acid 12% creams or lotions for itching and urea creams for dry skin. Oral and topical ketoconazole were found to result in a 90% reduction of cutaneous lesions after ten days of therapy [Liu et al 2015].
- Dermatologic surgery. An inflammatory nevus was removed from an affected boy by dermabrasion; however, it recurred within eight months [Happle et al 1996]. König et al [2010] reported successful treatment of an inflammatory nevus by grafting skin obtained from a contralateral unaffected region.
- Oral aromatic retinoids (etretinate) to ameliorate cutaneous symptoms; however, this drug is often poorly tolerated [Happle et al 1980] and does not prove effective in every individual [Liu et al 2015].
- Topical statins. The use of lovastatin topically led to complete healing of the inflammatory CHILD nevus in a few individuals, whereas cholesterol application alone had no satisfactory effect [Merino De Paz et al 2011, Paller et al 2011]. Alexopoulos & Kakourou [2015] reported the combined topical use of simvastatin and cholesterol and showed a correction in the cutaneous phenotype of one individual. The addition of glycolic acid to cholesterol and lovastatin creams improved the penetrance of this therapy into the thick skin scales, thus improving treatment [Bergqvist et al 2018]. Bajawi et al [2018] showed remarkable improvement of the skin lesions in one individual in response to treatment with simvastatin 2% ointment monotherapy.
- Note: No one therapy described to date appears to ameliorate the cutaneous findings for every reported individual with CHILD syndrome. Trying different methods until the clinician finds a successful therapy appears to be typical for most affected individuals.
- Orthopedic abnormalities. Treatment of orthopedic abnormalities such as scoliosis and joint contractures with braces and/or corrective surgery
- Other medical care as appropriate based on clinical findings
CK syndrome
- Behavior modification and/or drug therapy to control aggression and help with ADHD symptoms
- Anti-seizure medication to control seizures
- Ophthalmologic management of ocular abnormalities
Surveillance
CHILD syndrome
- Regular surveillance for cutaneous manifestations as new lesions may occur in puberty or early adulthood
- Orthopedic surveillance for musculoskeletal deformities such as scoliosis and joint contractures
- Neurologic, cardiologic, or renal surveillance depending on clinical involvement
CK syndrome
- Neurologic surveillance of seizures for readjustment of medications if necessary
- Orthopedic surveillance for scoliosis/kyphosis
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
The NSDHL-related disorders are inherited in an X-linked manner.
- CHILD syndrome is usually lethal to males during gestation and predominantly affects females.
- CK syndrome predominantly affects males.
Risk to Family Members ‒ CHILD Syndrome
Parents of a proband
- A female with CHILD syndrome may have inherited an NSDHL pathogenic variant from her mother or the pathogenic variant may be de novo. Theoretically, an affected female may also have inherited the variant from a father with germline mosaicism.
- Although CHILD syndrome-associated NSDHL variants appear to be highly penetrant, mothers heterozygous for an NSDHL pathogenic variant who have only mild skin lesions, Blaschko-linear inflammatory scaly lesions, patchy alopecia, and nail changes have been reported [Bittar et al 2006]. Favorably skewed X-chromosome inactivation has been proposed to explain this milder phenotype, and theoretically could on occasion result in a phenotypically normal heterozygous female. Therefore, an apparently negative family history cannot be confirmed unless molecular genetic testing has been performed on the mother of the proband.
- The father of an affected male will not have the disorder nor will he be hemizygous for an NSDHL pathogenic variant.
Sibs of a female proband. The risk to sibs depends on the genetic status of the mother.
- If the mother of the proband has an NSDHL pathogenic variant, the chance of transmitting it in each pregnancy is 50%. However, since studies suggest that male conceptuses with an NSDHL pathogenic variant generally abort or resorb spontaneously [Cunningham et al 2005], the expected live-born distribution is 33% unaffected females, 33% affected females, and 33% unaffected males.
- If the proband represents a simplex case (i.e., a single affected family member) and if pathogenic variant identified in the proband cannot be detected in the leukocyte DNA of the mother, the risk to sibs is slightly greater than that of the general population (though still <1%) because of the possibility of maternal germline mosaicism.
Offspring of a female proband
- The risk to the offspring of females with CHILD syndrome must take into consideration the presumed lethality to affected males during gestation.
- At conception, the chance of transmitting the pathogenic variant in each pregnancy is 50%; however, since male conceptuses with an NSDHL pathogenic variant generally abort or resorb spontaneously, the expected live-born distribution is 33% unaffected females, 33% affected females, and 33% unaffected males.
Other family members. The risk to other family members depends on the status of the proband's mother: if the proband's mother has the NSDHL pathogenic variant, her family members may be at risk.
Risk to Family Members ‒ CK Syndrome
Parents of a male proband
- The father of a male with CK syndrome will not have the disorder nor will he be hemizygous for the NSDHL pathogenic variant; therefore, he does not require further evaluation/testing.
- In a family with more than one affected individual, the mother of an affected male is an obligate heterozygote. Note: If a woman has more than one affected child and no other affected relatives and if the NSDHL pathogenic variant cannot be detected in her leukocyte DNA, she most likely has germline mosaicism. The frequency of germline mosaicism is unknown.
- If a male is the only affected family member (i.e., a simplex case), the mother may be a heterozygote or the affected male may have a de novo NSDHL pathogenic variant, in which case the mother is not a heterozygote. The frequency of de novo pathogenic variants is unknown.
Sibs of a male proband. The risk to sibs depends on the genetic status of the mother:
- If the mother of the proband is heterozygous for an NSDHL pathogenic variant, the expected chance of transmitting it in each pregnancy is 50%. However, in the three families reported to date, Preiksaitiene et al [2015] observed apparent preferential transmission of the NSDHL pathogenic variant: the transmission rate was approximately 82% (versus the expected 50% transmission rate). More families will have to be identified and analyzed to further substantiate this observation.
- Males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will usually not be affected (see Clinical Description, CK Syndrome, Heterozygous females).
- If the proband represents a simplex case and if the pathogenic variant cannot be detected in the leukocyte DNA of the mother, the risk to sibs is slightly greater than that of the general population (though still <1%) because of the possibility of maternal germline mosaicism. The frequency of germline mosaicism is unknown.
Offspring of a male proband. To date, no male with CK syndrome has reproduced.
Other family members. The proband's maternal aunts may be at risk of being heterozygotes for the pathogenic variant, and the aunts' offspring, depending on their sex, may be at risk of being heterozygotes for the pathogenic variant or of being affected.
Heterozygote Detection
Molecular genetic testing of at-risk female relatives to determine their genetic status is most informative if the pathogenic variant has been identified in the proband.
Note: Heterozygous females may have a range of clinical manifestations (see Clinical Description, CK Syndrome, Heterozygous females).
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of genetic status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, heterozygous, or at risk of being heterozygous.
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the NSDHL pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing for an NSDHL-related disorder are possible.
Biochemical testing is theoretically possible, although it has not been reported for NSDHL-related disorders. Additionally, limb deficiency or another skeletal anomaly detected by fetal ultrasound suggests the possibility of recurrence; however, mild manifestations may not have detectable limb findings.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- FIRST - Foundation for Ichthyosis and Related Skin TypesPhone: 215-997-9400; 800-545-3286Email: info@firstskinfoundation.org
- Rare Disease FoundationCanadaEmail: families@rarediseasefoundation.org; info@rarediseasefoundation.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
NSDHL-Related Disorders: Genes and Databases
Table B.
OMIM Entries for NSDHL-Related Disorders (View All in OMIM)
Gene structure. NSDHL comprises eight exons. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants
- CHILD syndrome. Well-studied CHILD-associated NSDHL variants result in loss of function of the protein and many are null alleles. Approximately 21 different pathogenic variants have been reported [König et al 2000, Saito & Ishiko 2008, Schmidt-Sidor et al 2008, Avgerinou et al 2010, Mi et al 2015]; they include missense, nonsense, frameshift, and splice site variants and deletions of all or portions of the gene [Bornholdt et al 2005, Kim et al 2005]. The following pathogenic variants have been seen more than once: p.Gly205Ser, p.Tyr349Cys, and p.Ala105Val.
- CK syndrome. CK syndrome-associated variants cause partial loss of enzyme function. The two pathogenic variants identified to date are a 3-bp in-frame deletion [McLarren et al 2010] and a pathogenic frameshift variant altering the carboxyl terminus of NSDHL [Tarpey et al 2009]. Both are temperature-sensitive pathogenic variants that decrease protein stability at 37º C [McLarren et al 2010]. Recently, a p.Gly152Asp missense variant was shown to segregate with CK syndrome in a Lithuanian family [Preiksaitiene et al 2015].
Table 6.
NSDHL Pathogenic Variants Discussed in This GeneReview
Normal gene product. The NSDHL enzyme, which localizes to the surface of the endoplasmic reticulum and to lipid droplets, is a member of a multiprotein complex and functions as a C4 demethylase in post-squalene cholesterol biosynthesis [Gachotte et al 1998, Mo et al 2002, Caldas & Herman 2003]. The protein contains 362 amino acids.
Abnormal gene product
- In CHILD syndrome, NSDHL pathogenic variants result in substantial or complete loss of functional NSDHL protein. Some pathogenic variants result in deletion of all or portions of NSDHL protein, whereas pathogenic nonsense and frameshift variants are likely to be loss-of-function variants [Rebbapragada & Lykke-Andersen 2009, McLarren et al 2010]. CHILD syndrome-associated missense variants studied by Lucas et al [2003] fail to complement in the yeast complementation system.
- In CK syndrome, NSDHL pathogenic variants result in severely reduced steady-state levels of NSDHL protein and thus lead to a partial loss of NSDHL protein function. In yeast complementation studies, these pathogenic variants complement as well as the wild type human NSDHL protein because the mutated proteins are stable at 30º C [McLarren et al 2010].
Chapter Notes
Author History
Cornelius F Boerkoel, MD, PhD (2010-present)
Christèle du Souich, MSc, CCGC, CGC (2010-present)
Karl-Heinz Grzeschik, PhD (2010-present)
Arne König, MD; Philipps-Universität, Marburg (2010-2015)
F Lucy Raymond, MD, PhD (2010-present)
Revision History
- 25 October 2018 (sw) Comprehensive update posted live
- 25 November 2015 (me) Comprehensive update posted live
- 27 June 2013 (me) Comprehensive update posted live
- 16 February 2012 (cd) Revision: deletion/duplication analysis for NSDHL available clinically
- 1 February 2011 (me) Review posted live
- 9 January 2010 (cb) Original submission
References
Literature Cited
- Alexopoulos A, Kakourou T. CHILD syndrome: successful treatment of skin lesions with topical simvastatin/cholesterol ointment--a case report. Pediatr Dermatol. 2015;32:e145–7. [PubMed: 25845514]
- Avgerinou GP, Asvesti AP, Katsambas AD, Nikolaou VA, Christofidou EC, Grzeschik KH, Happle R. CHILD syndrome: the NSDHL gene and its role in CHILD syndrome, a rare hereditary disorder. J Eur Acad Dermatol Venereol. 2010;24:733–6. [PubMed: 19906044]
- Baden HP, Rex IH Jr. Linear ichthyosis associated with skeletal abnormalities. Arch Dermatol. 1970;102:126–8. [PubMed: 4322292]
- Bajawi SM, Jafarri SA, Buraik MA, Al Attas KM, Hannani HY. Pathogenesis-based therapy: cutaneous abnormalities of CHILD syndrome successfully treated with topical simvastatin monotherapy. JAAD Case Rep. 2018;4:232–4. [PMC free article: PMC5909487] [PubMed: 29687057]
- Bergqvist C, Abdallah B, Hasbani DJ, Abbas O, Kibbi AG, Kurban M, Rubeiz N. CHILD syndrome: a modified pathogenesis-targeted therapeutic approach. Am J Med Genet A. 2018;176:733–8. [PubMed: 29392821]
- Bittar M, Happle R. CHILD syndrome avant la lettre. J Am Acad Dermatol. 2004;50:S34–7. [PubMed: 14726863]
- Bittar M, Happle R, Grzeschik KH, Leveleki L, Hertl M, Bornholdt D, König A. CHILD syndrome in 3 generations: the importance of mild or minimal skin lesions. Arch Dermatol. 2006;142:348–51. [PubMed: 16549711]
- Bornholdt D, König A, Happle R, Leveleki L, Bittar M, Danarti R, Vahlquist A, Tilgen W, Reinhold U, Poiares Baptista A, Grosshans E, Vabres P, Niiyama S, Sasaoka K, Tanaka T, Meiss AL, Treadwell PA, Lambert D, Camacho F, Grzeschik KH. Mutational spectrum of NSDHL in CHILD syndrome. J Med Genet. 2005;42:e17. [PMC free article: PMC1735983] [PubMed: 15689440]
- Bruno DL, Anderlid BM, Lindstrand A, van Ravenswaaij-Arts C, Ganesamoorthy D, Lundin J, Martin CL, Douglas J, Nowak C, Adam MP, Kooy RF, Van der Aa N, Reyniers E, Vandeweyer G, Stolte-Dijkstra I, Dijkhuizen T, Yeung A, Delatycki M, Borgström B, Thelin L, Cardoso C, van Bon B, Pfundt R, de Vries BB, Wallin A, Amor DJ, James PA, Slater HR, Schoumans J. Further molecular and clinical delineation of co-locating 17p13.3 microdeletions and microduplications that show distinctive phenotypes. J Med Genet. 2010;47:299–311. [PubMed: 20452996]
- Caldas H, Herman GE. NSDHL, an enzyme involved in cholesterol biosynthesis, traffics through the Golgi and accumulates on ER membranes and on the surface of lipid droplets. Hum Mol Genet. 2003;12:2981–91. [PubMed: 14506130]
- Chander R, Varghese B, Jabeen M, Garg T, Jain M. CHILD syndrome with thrombocytosis and congenital dislocation of the hip: a case report from India. Dermatol Online J. 2010;16:6. [PubMed: 20804683]
- Cunningham D, Swartzlander D, Liyanarachchi S, Davuluri RV, Herman GE. Changes in gene expression associated with loss of function of the NSDHL sterol dehydrogenase in mouse embryonic fibroblasts. J Lipid Res. 2005;46:1150–62. [PubMed: 15805545]
- du Souich C, Chou A, Yin J, Oh T, Nelson TN, Hurlburt J, Arbour L, Friedlander R, McGillivray BC, Tyshchenko N, Rump A, Poskitt KJ, Demos MK, Van Allen MI, Boerkoel CF. Characterization of a new X-linked mental retardation syndrome with microcephaly, cortical malformation, and thin habitus. Am J Med Genet A. 2009;149A:2469–78. [PubMed: 19842190]
- du Souich C, McLarren K, Nowaczyk MJM, Whitman JC, Livesley J, Woodward TS, Steiner RD, Boerkoel CF. CK syndrome: another glimpse of neurodevelopmental regulation by cholesterol biosynthesis. Abstract 281. Charlotte, NC: Society for Inherited Metabolic Disorders Annual Meeting; 2012.
- Falek A, Heath CW Jr, Ebbin AJ, McLean WR. Unilateral limb and skin deformities with congenital heart disease in two siblings: a lethal syndrome. J Pediatr. 1968;73:910–3. [PubMed: 5696317]
- Gachotte D, Barbuch R, Gaylor J, Nickel E, Bard M. Characterization of the Saccharomyces cerevisiae ERG26 gene encoding the C-3 sterol dehydrogenase (C-4 decarboxylase) involved in sterol biosynthesis. Proc Natl Acad Sci U S A. 1998;95:13794–9. [PMC free article: PMC24900] [PubMed: 9811880]
- Grange DK, Kratz LE, Braverman NE, Kelley RI. CHILD syndrome caused by deficiency of 3beta-hydroxysteroid-delta8, delta7-isomerase. Am J Med Genet. 2000;90:328–35. [PubMed: 10710233]
- Happle R, Effendy I, Megahed M, Orlow SJ, Küster W. CHILD syndrome in a boy. Am J Med Genet. 1996;62:192–4. [PubMed: 8882402]
- Happle R, Koch H, Lenz W. The CHILD syndrome. Congenital hemidysplasia with ichthyosiform erythroderma and limb defects. Eur J Pediatr. 1980;134:27–33. [PubMed: 7408908]
- Happle R, Mittag H, Kuster W. The CHILD nevus: a distinct skin disorder. Dermatology. 1995;191:210–16. [PubMed: 8534939]
- Hashimoto K, Topper S, Sharata H, Edwards M. CHILD syndrome: analysis of abnormal keratinization and ultrastructure. Pediatr Dermatol. 1995;12:116–29. [PubMed: 7544893]
- Hebert AA, Esterly NB, Holbrook KA, Hall JC. The CHILD syndrome. Histologic and ultrastructural studies. Arch Dermatol. 1987;123:503–9. [PubMed: 3827283]
- Herman GE, Kratz L. Disorders of sterol synthesis: beyond Smith-Lemli-Opitz syndrome. Am J Med Genet C Semin Med Genet. 2012;160C:301–21. [PubMed: 23042573]
- Hummel M, Cunningham D, Mullett CJ, Kelley RI, Herman GE. Left-sided CHILD syndrome caused by a nonsense mutation in the NSDHL gene. Am J Med Genet A. 2003;122A:246–51. [PubMed: 12966526]
- Kim CA, Konig A, Bertola DR, Albano LM, Gattás GJ, Bornholdt D, Leveleki L, Happle R, Grzeschik KH. CHILD syndrome caused by a deletion of exons 6-8 of the NSDHL gene. Dermatology. 2005;211:155–8. [PubMed: 16088165]
- Knape RM, Gandhi KB, Tuli SY, Khuddus N. Optic nerve findings in CHILD syndrome. J Pediatr Ophthalmol Strabismus. 2010;47 Online:e1-3. [PubMed: 20886807]
- König A, Happle R, Bornholdt D, Engel H, Grzeschik KH. Mutations in the NSDHL gene, encoding a 3beta-hydroxysteroid dehydrogenase, cause CHILD syndrome. Am J Med Genet. 2000;90:339–46. [PubMed: 10710235]
- König A, Happle R, Fink-Puches R, Soyer HP, Bornholdt D, Engel H, Grzeschik KH. A novel missense mutation of NSDHL in an unusual case of CHILD syndrome showing bilateral, almost symmetric involvement. J Am Acad Dermatol. 2002;46:594–6. [PubMed: 11907515]
- König A, Skrzypek J, Loffler H, Oeffner F, Grzeschik KH, Happle R. Donor dominance cures CHILD nevus. Dermatology. 2010;220:340–5. [PubMed: 20389027]
- Liu T, Qian G, Wang XX, Zhang YG. CHILD syndrome: effective treatment of ichthyosiform naevus with oral and topical ketoconazole. Acta Derm Venereol. 2015;95:91–2. [PubMed: 24696032]
- Lucas ME, Ma Q, Cunningham D, Peters J, Cattanach B, Bard M, Elmore BK, Herman GE. Identification of two novel mutations in the murine Nsdhl sterol dehydrogenase gene and development of a functional complementation assay in yeast. Mol Genet Metab. 2003;80:227–33. [PubMed: 14567972]
- McLarren KW, Severson TM, du Souich C, Stockton DW, Kratz LE, Cunningham D, Hendson G, Morin RD, Wu D, Paul JE, An J, Nelson TN, Chou A, DeBarber AE, Merkens LS, Michaud JL, Waters PJ, Yin J, McGillivray B, Demos M, Rouleau GA, Grzeschik KH, Smith R, Tarpey PS, Shears D, Schwartz CE, Gecz J, Stratton MR, Arbour L, Hurlburt J, Van Allen MI, Herman GE, Zhao Y, Moore R, Kelley RI, Jones SJ, Steiner RD, Raymond FL, Marra MA, Boerkoel CF. Hypomorphic temperature-sensitive alleles of NSDHL cause CK syndrome. Am J Hum Genet. 2010;87:905–14. [PMC free article: PMC2997364] [PubMed: 21129721]
- Merino De Paz N, Rodriguez-Martin M, Contreras-Ferrer P, Garcia Bustinduy M, Gonzalez Perera I, Virgos Aller T, Martin Herrera A, Noda Cabrera A. Topical treatment of CHILD nevus and Sjögren-Larsson syndrome with combined lovastatin and cholesterol. Eur J Dermatol. 2011;21:1026–7. [PubMed: 21983059]
- Mi XB, Luo MX, Guo LL, Zhang TD, Qiu XW. CHILD syndrome: case report of a Chinese patient and literature review of the NAD[P]H steroid dehydrogense-like protein gene mutation. Pediatr Dermatol. 2015;32:e277–82. [PubMed: 26459993]
- Mo C, Valachovic M, Randall SK, Nickels JT, Bard M. Protein-protein interactions among C-4 demethylation enzymes involved in yeast sterol biosynthesis. Proc Natl Acad Sci U S A. 2002;99:9739–44. [PMC free article: PMC124998] [PubMed: 12119386]
- Paller AS, van Steensel MA, Rodriguez-Martín M, Sorrell J, Heath C, Crumrine D, van Geel M, Cabrera AN, Elias PM. Pathogenesis-based therapy reverses cutaneous abnormalities in an inherited disorder of distal cholesterol metabolism. J Invest Dermatol. 2011;131:2242–8. [PMC free article: PMC3193573] [PubMed: 21753784]
- Preiksaitiene E, Caro A, Benušienė E, Oltra S, Orellana C, Morkūnienė A, Roselló MP, Kasnauskiene J, Monfort S, Kučinskas V, Mayo S, Martinez F. A novel missense mutation in the NSDHL gene identified in a Lithuanian family by targeted next-generation sequencing causes CK syndrome. Am J Med Genet A. 2015;167:1342–8. [PubMed: 25900314]
- Rebbapragada I, Lykke-Andersen J. Execution of nonsense-mediated mRNA decay: what defines a substrate? Curr Opin Cell Biol. 2009;21:394–402. [PubMed: 19359157]
- Ryan C, Quinn S, McDermott M. Small intestine mucosal xanthoma in a patient with CHILD syndrome. J Clin Pathol. 2013;66:1094–5. [PubMed: 23969275]
- Saito M, Ishiko A. A novel silent mutation in the NSDHL gene causing CHILD syndrome as a result of aberrant splicing. Br J Dermatol. 2008;159:1204–6. [PubMed: 18764845]
- Schmidt-Sidor B, Obersztyn E, Szymańska K, Wychowski J, Mierzewska H, Wierzba-Bobrowicz T, Stepień T. Brain and cerebellar hemidysplasia in a case with ipsilateral body dysplasia and suspicion of CHILD syndrome. Folia Neuropathol. 2008;46:232–7. [PubMed: 18825599]
- Tang TT, McCreadie SR. Q--congenital hemidysplasia with ichthyosis. Birth Defects Orig Artic Ser. 1974;10:257–61. [PubMed: 4620143]
- Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, O'Meara S, Latimer C, Dicks E, Menzies A, Stephens P, Blow M, Greenman C, Xue Y, Tyler-Smith C, Thompson D, Gray K, Andrews J, Barthorpe S, Buck G, Cole J, Dunmore R, Jones D, Maddison M, Mironenko T, Turner R, Turrell K, Varian J, West S, Widaa S, Wray P, Teague J, Butler A, Jenkinson A, Jia M, Richardson D, Shepherd R, Wooster R, Tejada MI, Martinez F, Carvill G, Goliath R, de Brouwer AP, van Bokhoven H, Van Esch H, Chelly J, Raynaud M, Ropers HH, Abidi FE, Srivastava AK, Cox J, Luo Y, Mallya U, Moon J, Parnau J, Mohammed S, Tolmie JL, Shoubridge C, Corbett M, Gardner A, Haan E, Rujirabanjerd S, Shaw M, Vandeleur L, Fullston T, Easton DF, Boyle J, Partington M, Hackett A, Field M, Skinner C, Stevenson RE, Bobrow M, Turner G, Schwartz CE, Gecz J, Raymond FL, Futreal PA, Stratton MR. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535–43. [PMC free article: PMC2872007] [PubMed: 19377476]
- Thevenon J, Callier P, Poquet H, Bache I, Menten B, Malan V, Cavaliere ML, Girod JP, Thauvin-Robinet C, El Chehadeh S, Pinoit JM, Huet F, Verges B, Petit JM, Mosca-Boidron AL, Marle N, Mugneret F, Masurel-Paulet A, Novelli A, Tümer Z, Loeys B, Lyonnet S, Faivre L. 3q27.3 microdeletion syndrome: a recognisable clinical entity associating dysmorphic features, marfanoid habitus, intellectual disability and psychosis with mood disorder. J Med Genet. 2014;51:21–7. [PubMed: 24133203]
- Wiedemeyer K, Hartschuh W. Trichoblastomas with Merkel cell proliferation in nevi sebacei in Schimmelpenning-Feuerstein-Mims syndrome--histological differentiation between trichoblastomas and basal cell carcinomas. J Dtsch Dermatol Ges. 2009;7:612–5. [PubMed: 19192012]
- Yu X, Zhang J, Gu Y, Wu Z, Bao L, Li M, Yao Z. CHILD syndrome mimicking verrucous nevus in a Chinese patient responded well to topical therapy of compound of simvastatin and cholesterol. JEADV. 2018;32:1209–13. [PubMed: 29341259]
- Zellweger H, Uehlinger E. A case of unilateral osteo-enchondromatosis (Ollier disease) with Naevus ichthyosiformis (article in German). Helv Paediatr Acta. 1948;3:153–63. [PubMed: 18865122]
Publication Details
Author Information and Affiliations
Vancouver, British Columbia, Canada
Cambridge, United Kingdom
Marburg, Germany
Vancouver, British Columbia, Canada
Publication History
Initial Posting: February 1, 2011; Last Update: October 25, 2018.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
du Souich C, Raymond FL, Grzeschik KH, et al. NSDHL-Related Disorders. 2011 Feb 1 [Updated 2018 Oct 25]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

