Summary
Clinical characteristics.
Myhre syndrome is a multisystem connective tissue disorder involving the skin and the cardiovascular, respiratory, gastrointestinal, and musculoskeletal systems. Affected individuals may experience progressive and proliferative fibrosis. Fibrosis may occur spontaneously or following trauma, invasive medical procedures, or surgery, often resulting in significant complications. Characteristic facial features are found in almost all affected individuals and are more apparent in older children and adults. Cardiovascular issues can include aortic hypoplasia and stenosis, congenital heart defects, pericardial involvement, and restrictive cardiomyopathy. Joint limitations may progress with age and resemble mild joint contractures. Most individuals have developmental delay / cognitive impairment, typically in the mild-to-moderate range. Other findings may include autism spectrum disorder, conductive or mixed hearing loss, short stature, refractive errors, premature puberty, recurrent respiratory infections, mechanical respiratory issues (choanal stenosis, laryngeal narrowing), and stenosis of the upper gastrointestinal tract.
Diagnosis/testing.
The diagnosis of Myhre syndrome is established in a proband with characteristic clinical findings and a heterozygous pathogenic (or likely pathogenic) variant in SMAD4 detected by molecular genetic testing.
Management.
Treatment of manifestations: Feeding therapy with a low threshold for a clinical and/or radiographic swallowing study; referral to nutrition for poor weight gain or obesity; medical therapy for systemic and/or pulmonary hypertension; long-term tracheostomy may be required for those with complete or recurrent tracheal stenosis; aggressive medical management for constipation; physical therapy to keep joints mobile; hearing aids may be helpful for those with hearing loss; some keloids can be treated with intralesional steroids with minimal invasiveness for lesion removal; and standard treatment for orofacial clefting / velopharyngeal insufficiency, congenital heart defects / pericardial disease, restrictive lung disease, gastrointestinal stenosis, developmental delay / intellectual disability, refractive errors / strabismus / cataracts, persistent middle ear effusions, immunodeficiency, diabetes mellitus, and pubertal/menstrual irregularities. Note: Growth hormone therapy for short stature is not currently recommended for individuals with Myhre syndrome.
Prevention of secondary complications: Limiting tissue trauma appears to be the single most important preventive measure: the literature suggests increased risk of proliferative fibrosis following otherwise uncomplicated endotracheal intubation and surgical procedures. When possible, alternative noninvasive approaches should be pursued during diagnosis and management.
Surveillance: At each visit: measure growth parameters, blood pressure, and oxygen pulse oximetry; monitor for respiratory insufficiency, signs/symptoms of upper-airway stenosis, constipation, developmental issues, behavioral issues, physical skills and mobility issues, premature puberty (in children), and frequent and/or unusual infections. Annually: perform pulmonary function studies (in children age >6 years who are able to cooperate), ophthalmology evaluation, and audiology evaluation. In asymptomatic individuals with a normal echocardiogram, repeat the echocardiogram every one to three years. Starting in the second decade: low threshold for fasting blood sugar and hemoglobin A1c to assess for diabetes; periodic DXA scan to monitor bone mineral density; monitor females with the c.1486C>T (p.Arg496Cys) pathogenic variant for menstrual irregularities.
Genetic counseling.
Myhre syndrome is inherited in an autosomal dominant manner. Most probands with Myhre syndrome have the disorder as a result of a de novo SMAD4 pathogenic variant; however, vertical transmission from a parent to child has been rarely observed. If the SMAD4 pathogenic variant found in the proband cannot be detected in the leukocyte DNA of either parent, the risk to sibs is presumed to be slightly greater than that of the general population (though still <1%) because of the theoretic possibility of parental germline mosaicism. Once the SMAD4 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at theoretic increased risk for Myhre syndrome and preimplantation genetic testing are possible.
Diagnosis
Formal clinical diagnostic criteria for Myhre syndrome have not been published.
Suggestive Findings
Myhre syndrome should be suspected in individuals with the following clinical, imaging, and family history findings. Although no single feature is pathognomonic, co-occurrence of some is highly suggestive of Myhre syndrome.
Clinical Findings
Suggestive clinical findings include the following:
- Short stature (height is significantly less than that predicted by parental heights) with compact body habitus in most affected individuals
- Characteristic facial features (See Clinical Description, Craniofacial Features.)
- Conductive and mixed hearing loss
- Respiratory difficulties, usually due to restrictive pulmonary disease; in rare cases due to laryngotracheal narrowing (including subglottic stenosis) or choanal stenosis
- Progressively stiff and thickened skin
- Limited range of motion of the joints
- Effusions involving the serosal surfaces of the heart (pericarditis), lungs, and peritoneum, which may progress to fibrosis
- Mild-to-moderate intellectual disability
- Autism or neurodivergent behaviors
- Severe constipation due to intestinal strictures
- Premature puberty
Imaging Findings
Echocardiographic findings
- Aortic narrowing, such as typical juxtaductal aortic coarctation, diffuse long-segment aortic hypoplasia, or segmental stenosis (branch arteries)
- Congenital heart defects including tetralogy of Fallot and obstructive defects of the left heart (aortic stenosis, mitral stenosis)
- Pericardial involvement ranging from transient effusion to chronic severe constrictive pericarditis
- Restrictive cardiomyopathy
- Pulmonary hypertension
Skeletal radiographs (See Figure 7.)
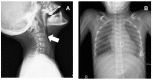
Figure 7.
Radiographs of a female with Myhre syndrome at age 14 years A. Lateral cervical spine shows thickened calvaria and anterior cervical vertebral fusion (arrow) of C2 and C3.
- Thickened calvarium
- Shortened long bones
- Enlarged vertebrae with shortened pedicles
- Cervical vertebral fusion
- Hypoplastic iliac wings
- Absent or extra ribs
Family History
Because Myhre syndrome is typically caused by a de novo pathogenic variant, most probands represent a simplex case (i.e., a single occurrence in a family). Rarely, the family history may be consistent with autosomal dominant inheritance (e.g., affected males and females in multiple generations).
Establishing the Diagnosis
The diagnosis of Myhre syndrome is established in a proband with characteristic clinical findings and a heterozygous pathogenic (or likely pathogenic) variant in SMAD4 detected by molecular genetic testing (see Table 1).
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include any likely pathogenic variants. (2) Identification of a heterozygous SMAD4 variant of uncertain significance does not establish or rule out the diagnosis.
Molecular genetic testing approaches can include a combination of gene-targeted testing (single-gene testing, multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those in whom the diagnosis of Myhre syndrome has not been considered are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
When the phenotypic and radiographic findings suggest the diagnosis of Myhre syndrome, molecular genetic testing approaches can include single-gene testing or use of a multigene panel:
- Single-gene testing. Sequence analysis of SMAD4 is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions.
- An autism/intellectual disability, hearing loss, or cancer multigene panel that includes SMAD4 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Option 2
When the diagnosis of Myhre syndrome has not been considered, genomic testing may be used. Comprehensive genomic testing does not require the clinician to determine which gene is likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Myhre Syndrome
Clinical Characteristics
Clinical Description
Myhre syndrome is a multisystem connective tissue disorder involving the cardiovascular system, respiratory system, gastrointestinal tract, musculoskeletal system, and skin. Affected individuals may experience progressive and proliferative fibrosis that may occur spontaneously or following trauma, invasive medical procedures, or surgery, often resulting in significant complications.
To date, more than 100 affected individuals with a molecularly confirmed diagnosis of Myhre syndrome have been reported [Le Goff et al 2011, Al Ageeli et al 2012, Asakura et al 2012, Caputo et al 2012, Lindor et al 2012, Picco et al 2013, Ishibashi et al 2014, Kenis et al 2014, Michot et al 2014, Hawkes & Kini 2015, Oldenburg et al 2015, Starr et al 2015, Bassett et al 2016, Lin et al 2016, Lin et al 2020, Cappuccio et al 2021, Cappuccio et al 2022, Yang et al 2022]. The following descriptions of the phenotypic features associated with Myhre syndrome are based on these reports.
Table 2.
Myhre Syndrome: Frequency of Select Features
Craniofacial Features
The craniofacial features of Myhre syndrome (Figures 1-6) can vary considerably and include:
- Short palpebral fissures (See Figure 4.)
- Deeply set eyes
- Maxillary underdevelopment
- Short philtrum
- Narrow mouth
- Thin vermilion of the upper lip (See Figure 3.)
- Small and/or widely spaced teeth
- Prognathism

Figure 4.
Female with Myhre syndrome at age five years. Note the short palpebral fissures, thin vermilion of the upper and lower lips, left-sided facial palsy, and brachydactyly, with otherwise mild features. Reported by Hawkes & Kini [2015]

Figure 3.
Male with Myhre syndrome at age 12 years with mild facial features (mild maxillary underdevelopment and thin vermilion of the upper lip) and finger contractures. Reported as Patient 4 in Starr et al [2015]
While cleft lip and palate is rare, velopharyngeal insufficiency is common.
Progression of Findings
The facial characteristics can progress over time. In infancy, the characteristic facial features are usually present but more difficult to recognize than in an older child (see Figures 1, 2, 5, and 6). Although classic coarsening of features is not present, mandibular elongation is notable. In most, short stature and hearing loss develop over time. The other highly distinctive (and often severe) findings of Myhre syndrome (joint stiffness, restrictive lung and cardiovascular disease, progressive and proliferative fibrosis, and thickening of the skin) also develop over time.



Figure 5.
The same male with Myhre syndrome at various ages from toddlerhood (lower right corner) to age 19 years (upper left corner).

Figure 6.
Two different women with Myhre syndrome at ages 40 years (A) and 50 years (B). The women are shown together in C.
Cardiovascular Features
Progressive cardiovascular issues can appear at any age; those with onset in childhood may worsen following instrumentation. Two affected individuals with restrictive cardiomyopathy who were treated with heart and heart/lung transplantation did not survive postoperative complications [Starr et al 2015].
In 54 individuals with confirmed Myhre syndrome [Lin et al 2016], 70% had a cardiovascular abnormality including structural heart defects (63%), pericardial disease (17%), restrictive cardiomyopathy (9%), and systemic hypertension (15%).
Congenital cardiovascular abnormalities include the following:
- Atrial septal defect or ventricular septal defect
- Patent ductus arteriosus
- Tetralogy of Fallot
- Obstructive defects of the left heart, such as juxtaductal aortic coarctation, long-segment aortic narrowing, aortic valve stenosis, and mitral valve stenosis. These are more common than obstructive defects of the right side, such as valvar and branch pulmonary artery stenosis [Michot et al 2014, Hawkes & Kini 2015, Starr et al 2015].
- Visceral vascular stenoses (in celiac, superior mesenteric, inferior mesenteric, and/or renal arteries)
Pericardial disease can present as short-term or recurrent effusions, or as chronic or progressive constrictive pericarditis that may require surgical intervention (see Management).
- Restrictive cardiomyopathy, a lethal condition, can be difficult to diagnose without cardiac catheterization.
- While constrictive pericarditis and restrictive cardiomyopathy can present with similar hemodynamic impairment, they differ in their pathogenesis and treatment (see Management).
Pulmonary hypertension, either primary or as a result of left ventricular dysfunction, has been infrequently reported; however, this may reflect limited evaluation and/or bias toward ascertainment and/or reporting of younger affected individuals (as underlying causes of pulmonary hypertension resulting from involvement of the lungs and cardiovascular circulation may evolve with age). It is unknown how often this is secondary to right-sided cardiac dysfunction, or severe left-sided obstruction, although both have been observed [Yang et al 2022].
Skeletal
Reduced range of motion of large and small joints is characteristic of Myhre syndrome and is exacerbated with age. Posture may be distinct with flexed elbows and bending forward at the hips (see Ishibashi et al [2014], Figure 1). Walking on tiptoes is common. Other features that may be present:
- Clinodactyly in more than half of affected individuals
- Syndactyly of the toes, usually 2-3
- Scoliosis
- Absence of normal lumbar lordosis and straight spine
- Sacral dimple, only rarely associated with a tethered spinal cord
Characteristic radiographic findings in affected individuals are listed in Suggestive Findings.
Developmental Delay and Intellectual Disability
Mild-to-moderate intellectual disability and developmental delay are common; however, cognition can be within the normal range. Delayed speech can be significant. Of note, acquired and unrecognized hearing loss may also contribute to speech delay and academic and social challenges. Affected adults have been employed and have married and reproduced.
Other Neurodevelopmental Features
Autism spectrum disorder has been noted in most affected individuals [Yang et al 2022].
Growth
The majority of affected infants have intrauterine growth restriction (IUGR). Short stature with compact body habitus (with normal head circumference) becomes more apparent over time.
- Most affected individuals have short long bones.
- Adult height is expected to be more than two standard deviations below what is predicted by parental heights in more than 80% of affected individuals, particularly in those who have a pathogenic variant at codon position 500 (see Genotype-Phenotype Correlations).
- Although head circumference is rarely greater than or equal to two standard deviations above the mean for age and sex, it is commonly proportionally greater than height ("relative macrocephaly").
- Some infants and children have difficulty with poor feeding and weight gain and may benefit from a feeding tube.
- Overweight (BMI >25, or >99th centile) may begin in adolescence, and is found in most adults.
Hearing Loss
Hearing loss is observed in most individuals with Myhre syndrome. Most newborns pass their neonatal hearing screen, and hearing loss usually becomes evident in early childhood to late teens.
- Hearing loss is predominantly conductive or mixed.
- Inner ear anomalies are rare.
- The underlying etiology of the hearing loss is often unclear or unknown; affected individuals most often have a history of bilateral myringotomy tube placement.
Primary Cutaneous Findings
Thick, firm skin is seen in nearly all individuals with Myhre syndrome, and stiffness may progress in many adults. Various terms used to describe the skin include thick, stiff, firm, rough, hyperkeratosis, and inelastic. Skin changes may not be apparent in infancy; they are often first noted on the extensor surfaces, palms, and soles. The changes progress with age. Additional skin findings include minimal creasing of the facial skin and unusual white linear scars.
Fibrosis
Proliferative fibrosis / abnormal scarring can occur following trauma or surgery. Some individuals develop hypertrophic, keloid-like scars. In addition to the skin, proliferation can also involve the large airways (trachea and bronchi) and the serosal surfaces of the heart, lungs, and peritoneum.
Respiratory Findings
Respiratory issues can be multifactorial. Laryngotracheal narrowing (including subglottic stenosis) may be congenital rather than post-traumatic. Interestingly, many children have had numerous intubations without developing "traumatic" stenosis. This can be mild in childhood, and rarely progresses to a severe multilevel form. Less common is upper-airway obstruction caused by choanal stenosis, which can rarely progress to complete stenosis (atresia).
Other findings can include the following:
- Restrictive pulmonary disease has been reported, often associated with restrictive thorax.
- "Asthma" may be diagnosed but does not always respond to bronchodilator therapy, as in typical reactive airway disease.
- Interstitial lung disease has been described. Severe pulmonary fibrosis has been noted on autopsy [Starr et al 2015, Starr et al 2022].
- Abnormal sleep is usually associated with autism. In some instances, a sleep study may reveal obstructive sleep apnea.
Immune System
Recurrent infections (especially otitis media and pneumonia) are common.
- Increased frequency of respiratory infections have been reported and may result from mechanical factors. For example, ear canals, sinuses, and mastoid cells may be opacified from proliferative debris.
- Serum immunoglobulin deficiency was detected in three affected individuals.
- Intravenous immunoglobulin was utilized with reported benefit in one affected individual [Starr et al 2015].
- It is unknown if immune deficiency is associated with Myhre syndrome or if it is an incidental finding [Michot et al 2014, Starr et al 2015].
Ophthalmologic Findings
Refractive errors are common and usually include hyperopia with astigmatism. Other findings may include strabismus, cataracts, corectopia, and optic nerve anomalies.
Endocrine Findings
Premature puberty may occur in both sexes. There can be early menarche, meno/metrorrhagia, and macromastia, the latter prompting reduction mammoplasty. Both premature ovarian failure and secondary amenorrhea have been observed.
Insulin-dependent diabetes mellitus has been noted in older adults.
Gastrointestinal Findings
Gastrointestinal involvement may include the following:
- Choking, coughing, and dysphagia
- Severe constipation
- Duodenal atresia
- Late-onset and congenital pyloric stenosis; less commonly stenosis may involve the duodenum and jejunum
- Protein-losing enteropathy associated with right heart failure and restrictive cardiomyopathy (Patient 1 in Lin et al [2016])
- Hirschsprung disease
Neoplasia
Since a report of neoplasia in six individuals with Myhre syndrome (3 of whom were women with endometrial cancer) [Lin et al 2020], there have been no additional known affected individuals with neoplasia.
Telangiectasias and juvenile polyps, reported in heterozygotes for a SMAD4 loss-of-function pathogenic variant, have not been reported in Myhre syndrome.
Hereditary hemorrhagic telangiectasia (HHT) and Myhre syndrome have opposing phenotypes consistent with SMAD4 loss of function and gain of function, respectively [Gheewalla et al 2022] (see Genetically Related Disorders).
Genotype-Phenotype Correlations
Genotype-phenotype correlations are still emerging.
c.1498A>G (p.Ile500Val). Based on limited data, individuals with the highly recurrent c.1498A>G (p.Ile500Val) pathogenic variant are more likely to have prenatal growth deficiency and postnatal short stature.
c.1486C>T (p.Arg496Cys). Individuals with the c.1486C>T (p.Arg496Cys) pathogenic variant are more likely to have a height within the normal range for age and sex. Females with this pathogenic variant are more likely to have premature puberty and heavy menses (see Management). Of the six individuals reported with neoplasia, three were women with endometrial cancer, two of whom were heterozygous for the c.1486C>T (p.Arg496Cys) pathogenic variant.
Nomenclature
LAPS (laryngotracheal stenosis, arthropathy, prognathism, and short stature) syndrome was determined to be a phenotypic variant of Myhre syndrome with pathogenic variants in the same codons [Lindor et al 2012, Picco et al 2013, Michot et al 2014]; the term is no longer in use.
Prevalence
The prevalence of Myhre syndrome is unknown. A rough estimate of the prevalence is 1:1,000,000 individuals.
Genetically Related (Allelic) Disorders
Other phenotypes known to be associated with germline pathogenic variants in SMAD4:
Whereas Myhre syndrome is caused by heterozygous gain-of-function SMAD4 pathogenic variants (see Molecular Genetics), JPS and HHT are caused by heterozygous loss-of-function SMAD4 pathogenic variants.
Differential Diagnosis
The disorders that most closely resemble Myhre syndrome are the other acromelic dysplasias – geleophysic dysplasia, acromicric dysplasia, and Weill-Marchesani syndrome – which share the findings of thickened skin, short stature, short hands, and stiff joints. Table 3 lists these and other syndromes that have more limited overlapping features.
Table 3.
Disorders to Consider in the Differential Diagnosis of Myhre Syndrome
Management
No clinical practice guidelines for Myhre syndrome have been published.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with Myhre syndrome, the evaluations summarized in Table 4 (if not completed previously as part of the diagnostic evaluation) are recommended.
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with Myhre Syndrome
Treatment of Manifestations
There is no cure for Myhre syndrome.
Supportive care to improve quality of life, maximize function, and reduce complications is recommended, ideally involving multidisciplinary care by specialists in relevant fields (see Table 5).
Table 5.
Treatment of Manifestations in Individuals with Myhre Syndrome
Developmental Delay / Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy as well as infant mental health services, special educators, and sensory impairment specialists. In the US, early intervention is a federally funded program available in all states that provides in-home services to target individual therapy needs.
Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (IEP) is developed for those who qualify based on established motor, language, social, or cognitive delay. The early intervention program typically assists with this transition. Developmental preschool is center based; for children too medically unstable to attend, home-based services are provided.
All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies (US) and to support parents in maximizing quality of life. Some issues to consider:
- IEP services:
- An IEP provides specially designed instruction and related services to children who qualify.
- IEP services will be reviewed annually to determine whether any changes are needed.
- Special education law requires that children participating in an IEP be in the least restrictive environment feasible at school and included in general education as much as possible, when and where appropriate.
- Vision and hearing consultants should be a part of the child's IEP team to support access to academic material.
- PT, OT, and speech services will be provided in the IEP to the extent that the need affects the child's access to academic material. Beyond that, private supportive therapies based on the affected individual's needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician.
- As a child enters the teen years, a transition plan should be discussed and incorporated in the IEP. For those receiving IEP services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, and modified assignments.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility and to reduce the risk for later-onset orthopedic complications (e.g., contractures).
- Consider use of durable medical equipment and positioning devices as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses, or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety.
Communication issues. Consider evaluation for alternative means of communication (e.g., augmentative and alternative communication [AAC]) for individuals who have expressive language difficulties. An AAC evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, AAC devices do not hinder verbal development of speech, but rather support optimal speech and language development.
Social/Behavioral Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). ABA therapy is targeted to the individual child's behavioral, social, and adaptive strengths and weaknesses and typically performed one-on-one with a board-certified behavior analyst.
Prevention of Secondary Complications
Limiting tissue trauma appears to be the single most important preventive measure: the literature suggests increased risk of proliferative fibrosis following otherwise uncomplicated endotracheal intubation and surgical procedures. When possible, alternative noninvasive approaches should be pursued during diagnosis and management [Oldenburg et al 2015, Starr et al 2015].
- Extreme care with intubation and use of an endotracheal tube without a cuff (or careful monitoring of pressures with a cuff) may help prevent airway stenosis [Oldenburg et al 2015].
- Minimize abdominal and pelvic procedures as extensive adhesions may develop postoperatively [Lindor et al 2012].
- Hysterectomy should be an option of last resort for treatment of menorrhagia as postsurgical fibrosis can occur.
- Recognize risk of thickened scars or keloids with ear/other piercing.
- Use of orthodontic braces may stimulate gum hypertrophy.
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations in Table 6 are recommended.
Table 6.
Recommended Surveillance for Individuals with Myhre Syndrome
Agents/Circumstances to Avoid
Affected individuals should be aggressively counseled not to smoke.
Limiting tissue trauma (injury) appears to be the single most important preventive concept in this disorder to communicate to all health care providers involved in individuals' care (see Prevention of Secondary Complications). Decision making with affected individuals and their families should include nonintervention as an option in, for example, ear piercing, orthodontic braces, or surgical repair of velopharyngeal insufficiency. Elective tracheal surgery/intubation should be avoided.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
The antihypertensive drug losartan is an angiotensin II type 1 receptor blocker. Through this mechanism, it also indirectly antagonizes transforming growth factor beta (TGF-β) signaling. In Myhre syndrome fibroblasts, losartan corrected an extracellular matrix deposition defect [Piccolo et al 2014]. Thus, in a small uncontrolled open-label pilot study, three individuals with Myhre syndrome were treated with losartan. Improvements in skin thickness, joint range of motion, and myocardial strain were observed [Cappuccio et al 2021]. However, long-term controlled clinical trials with a larger number of affected individuals are needed to establish the efficacy of losartan on skin, joint, and heart abnormalities in Myhre syndrome.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Myhre syndrome is an autosomal dominant disorder typically caused by a de novo pathogenic variant.
Risk to Family Members
Parents of a proband
- Most probands with Myhre syndrome have the disorder as the result of a de novo SMAD4 pathogenic variant [Lin et al 2016].
- Vertical transmission (from an affected mother to two affected children) has been reported in one family [Meerschaut et al 2019]. Additional families with vertical transmission have been identified [Author, personal observation].
- If the proband appears to be the only affected family member (i.e., a simplex case), molecular genetic testing is recommended for the parents of the proband to confirm their genetic status and to allow reliable recurrence risk counseling.
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism, which has not been reported to date in Myhre syndrome. Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ cells only.
Sibs of a proband. The risk to the sibs of the proband depends on the genetic status of the proband's parents:
- If the SMAD4 pathogenic variant identified in the proband cannot be detected in the leukocyte DNA of either parent, the recurrence risk to sibs is estimated to be 1% because of the theoretic possibility of parental germline mosaicism [Rahbari et al 2016].
- If a parent of the proband is affected and/or is known to have the pathogenic variant identified in the proband, the risk to the sibs of inheriting the pathogenic variant is 50%.
- If the parents have not been tested for the SMAD4 pathogenic variant but are clinically unaffected, the risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for Myhre syndrome because of the theoretic possibility of reduced penetrance in a heterozygous parent or parental germline mosaicism.
Offspring of a proband. Each child of an individual with Myhre syndrome has a 50% chance of inheriting the SMAD4 pathogenic variant.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent has the SMAD4 pathogenic variant, the parent's family members may be at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to parents of affected individuals.
Prenatal Testing and Preimplantation Genetic Testing
Once the SMAD4 pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing for Myhre syndrome are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- Myhre Syndrome Foundation
- National Organization for Rare Disorders (NORD)
- Alexander Graham Bell Association for the Deaf and Hard of HearingPhone: 866-337-5220 (toll-free); 202-337-5221 (TTY)Fax: 202-337-8314Email: info@agbell.org
- American Society for Deaf ChildrenPhone: 800-942-2732 (ASDC)Email: info@deafchildren.org
- National Association of the DeafPhone: 301-587-1788 (Purple/ZVRS); 301-328-1443 (Sorenson); 301-338-6380 (Convo)Fax: 301-587-1791Email: nad.info@nad.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Myhre Syndrome: Genes and Databases
Table B.
OMIM Entries for Myhre Syndrome (View All in OMIM)
Molecular Pathogenesis
Heterozygous gain-of-function pathogenic variants in SMAD4 confer stability of the resulting abnormal protein due to an apparent decrease in monoubiquitination. This affects transforming growth factor beta (TGF-β) signaling, thus altering expression of downstream target genes encoding TGF-β and bone morphogenic proteins (BMP), resulting in abnormal extracellular matrix deposition and altered development of the axial and appendicular skeleton, cardiac muscle, and central nervous system [Caputo et al 2012, Le Goff et al 2014, Piccolo et al 2014].
In contrast, heterozygosity for a loss-of-function SMAD4 pathogenic variant has been well established as the cause of a spectrum of acquired cardiac diseases, including cardiac fibrosis and hypertrophy, aortopathies, atherogenesis, and pulmonary artery hypertension [Andrabi et al 2011, Nasim et al 2011, Heald et al 2015].
Mechanism of disease causation. Gain of function had been suspected; however, new research suggests a dominant-negative mechanism causing an interruption of typical TGF and BMP signaling [Alankarage et al 2022].
Table 7.
Notable SMAD4 Pathogenic Variants
Cancer and benign tumors. Although germline SMAD4 loss-of-function (inactivating) pathogenic variants predispose to hamartomatous polyps in the gastrointestinal track (see Juvenile Polyposis Syndrome), the gain-of-function pathogenic variants associated with Myhre syndrome show no such associations (see Clinical Description, Neoplasia).
Note that somatic inactivation of SMAD4, a gastrointestinal malignancy-specific tumor suppressor gene, is found in one third of colorectal cancer specimens and half of pancreatic tumors [Chen et al 2014].
Chapter Notes
Author Notes
Myhre Syndrome Clinic at Massachusetts General Hospital
The Lindsay Lab at Massachusetts General Hospital
Acknowledgments
The authors are indebted to the people living with Myhre syndrome and their families who have provided consent, motivation, contributions, and advocacy.
Author History
Nicola Brunetti-Pierri, MD (2022-present)
Angela E Lin, MD (2017-present)
Noralane M Lindor, MD; Mayo Clinic (2017-2022)
Mark E Lindsay, MD, PhD (2022-present)
Lisa A Schimmenti, MD (2022-present)
Lois J Starr, MD, PhD (2017-present)
Revision History
- 24 November 2022 (ma) Comprehensive update posted live
- 13 April 2017 (bp) Review posted live
- 11 July 2016 (ljs) Original submission
References
Literature Cited
- Al Ageeli E, Mignot C, Afenjar A, Whalen S, Dorison N, Mayer M, Esteva B, Dubern B, Momtchilova M, Le Gargasson JF, Bursztyn J, Heron D. Retinal involvement in two unrelated patients with Myhre syndrome. Eur J Med Genet. 2012;55:541-7. [PubMed: 22683461]
- Alankarage D, Enriquez A, Steiner RD, Raggio C, Higgins M, Milnes D, Humphreys DT, Duncan EL, Sparrow DB, Giampietro PF, Chapman G, Dunwoodie SL. Myhre syndrome is caused by dominant-negative dysregulation of SMAD4 and other co-factors. Differentiation. 2022;128:1-12. [PMC free article: PMC10442510] [PubMed: 36194927]
- Andrabi S, Bekheirnia MR, Robbins-Furman P, Lewis RA, Prior TW, Potocki L. SMAD4 mutation segregating in a family with juvenile polyposis, aortopathy, and mitral valve dysfunction. Am J Med Genet A. 2011;155A:1165-9. [PubMed: 21465659]
- Asakura Y, Muroya K, Sato T, Kurosawa K, Nishimaura G, Adachi M. First case of a Japanese girl with Myhre syndrome due to a heterozygous SMAD4 mutation. Am J Med Genet Part A. 2012;158A:1982-6. [PubMed: 22711472]
- Bassett JK, Douzgou S, Kerr B. Severe constipation in a patient with Myhre syndrome: a case report. Clin Dysmorph. 2016;25:54-7. [PubMed: 26636501]
- Cappuccio G, Brunetti-Pierri N, Clift P, Learn C, Dykes JC, Mercer CL, Callewaert B, Meerschaut I, Spinelli AM, Bruno I, Gillespie MJ, Dorfman AT, Grimberg A, Lindsay ME, Lin AE. Expanded cardiovascular phenotype of Myhre syndrome includes tetralogy of Fallot suggesting a role for SMAD4 in human neural crest defects. Am J Med Genet A. 2022;188:1384-95. [PubMed: 35025139]
- Cappuccio G, Caiazza M, Roca A, Melis D, Iuliano A, Matyas G, Rubino M, Limongelli G, Brunetti-Pierri N. A pilot clinical trial with losartan in Myhre syndrome. Am J Med Genet A. 2021;185:702-9. [PMC free article: PMC7898344] [PubMed: 33369056]
- Caputo V, Cianetti L, Niceta M, Carta C, Ciolfi A, Bocchinfuso G, Carrani E, Dentici ML, Biamino E, Belligni E, Garavelli L, Boccone L, Melis D, Andria G, Gelb BD, Stella L, Silengo M, Dallapiccola B, Tartaglia M. A restricted spectrum of mutations in the SMAD4 tumor-suppressor gene underlies Myhre syndrome. Am J Hum Genet. 2012;90:161-9. [PMC free article: PMC3257749] [PubMed: 22243968]
- Chen YW, Hsiao PJ, Weng CC, Kuo KK, Kuo TL, Wu DC, Hung WC, Cheng KH. SMAD4 loss triggers the phenotypic changes of pancreatic ductal adenocarcinoma cells. BMC Cancer. 2014;14:181. [PMC free article: PMC4007528] [PubMed: 24625091]
- Garavelli L Maini I, Baccilieri F, Ivanovski I, Pollazzon M, Rosato S, Iughetti L, Unger S, Superti-Furga A, Tartaglia M. Natural history and life-threatening complications in Myhre syndrome and review of the literature. Eur J Pediatr. 2016;175:1307-15. [PubMed: 27562837]
- Gheewalla GM, Luther J, Das S, Kreher JB, Scimone ER, Wong AW, Lindsay ME, Lin AE. An additional patient with SMAD4-juvenile polyposis-hereditary hemorrhagic telangiectasia and connective tissue abnormalities: SMAD4 loss-of-function and gain-of-function pathogenic variants result in contrasting phenotypes. Am J Med Genet A. 2022;188:3084-8. [PubMed: 35869926]
- Hawkes L, Kini U. Myhre syndrome with facial paralysis and branch pulmonary stenosis. Clin Dysmorph. 2015;24:84-5. [PubMed: 25486016]
- Heald B, Rigelsky C, Moran R, LaGuardia L, O'Malley M, Burke CA, Zahka K. Prevalence of thoracic aortopathy in patients with juvenile polyposis syndrome-hereditary hemorrhagic telangiectasia due to SMAD4. Am J Med Genet A. 2015;167A:1758-62. [PubMed: 25931195]
- Ishibashi N, Sasaki Y, Asakura Y. Myhre syndrome: a rare craniofacial disorder. Cranio. 2014;32:300-6. [PubMed: 25252769]
- Kenis C, Verstreken M, Gieraerts K, De Foer B, Van der Aa N, Offeciers EF, Casselman JW. Bilateral otospongiosis and a unilateral vestibular schwannoma in a patient with Myhre syndrome. Otol Neurotol. 2014;35:e253-5. [PubMed: 24841914]
- Le Goff C, Mahaut C, Abhyankar A, Le Goff W, Serre V, Afenjar A, Destrée A, di Rocco M, Héron D, Jacquemont S, Marlin S, Simon M, Tolmie J, Veroles A, Casanova JL, Munnich A, Cormier-Daire V. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat Genet. 2011;44:85-8. [PubMed: 22158539]
- Le Goff C, Michot C, Cormier-Daire V. Myhre syndrome. Clin Genet. 2014;85:503-13. [PubMed: 24580733]
- Lin AE, Alali A, Starr LJ, Shah N, Beavis A, Pereira EM, Lindsay ME, Klugman S. Gain-of-function pathogenic variants in SMAD4 are associated with neoplasia in Myhre syndrome. Am J Med Genet A. 2020;182:328-37. [PubMed: 31837202]
- Lin AE, Michot C, Cormier-Daire V, L'Ecuyer TJ, Matherne GP, Barnes BH, Humberson JB, Edmondson AC, Zackai E, O'Connor MJ, Kaplan JD, Ebeid MR, Krier J, Krieg E, Ghoshhajra B, Lindsay ME. Gain-of-function mutations in SMAD4 cause a distinctive repertoire of cardiovascular phenotypes in patients with Myhre syndrome. Am J Med Genet A. 2016;170:2617-31. [PubMed: 27302097]
- Lindor NM, Gunawardena SR, Thibodeau SN. Mutations of SMAD4 account for both LAPS and Myhre syndromes. Am J Med Genet Part A. 2012;158A:1520-1. [PubMed: 22585601]
- McGowan R, Gulati R, McHenry P, Cooke A, Butler S, Teik Keng W, Murday V, Whiteford M, Dikkers FG, Sikkema-Raddatz B, van Essen T, Tolmie J. Clinical features and respiratory complications in Myhre syndrome. Eur J Med Genet. 2011;54:e553-e559. [PubMed: 21816239]
- Meerschaut I, Beyens A, Steyaert W, De Rycke R, Bonte K, De Backer T, Janssens S, Panzer J, Plasschaert F, De Wolf D, Callewaert B. Myhre syndrome: a first familial recurrence and broadening of the phenotypic spectrum. Am J Med Genet A. 2019;179:2494-9. [PubMed: 31595668]
- Michot C, Le Goff C, Mahaut C, Afenjar A, Brooks AS, Campeau PM, Destree A, Di Rocco M, Donnai D, Hennekam R, Heron D, Jacquemont S, Kannu P, Lin AE, Manouvrier-Hanu S, Mansour S, Marlin S, McGowan R, Murphy H, Raas-Rothchild A, Rio M, Simon M, Stolte-Dijkstra I, Stone JR, Szanjer Y, Tolmie J, Touraine R, Ende JVD, Van der Aa N, Essen TV, Verloes A, Munnich A, Comier-Daire V. Myhre and LAPS syndromes: clinical and molecular review of 32 patients. Eur J Hum Genet. 2014;22:1272-7. [PMC free article: PMC4200423] [PubMed: 24424121]
- Nasim MT, Ogo T, Ahmed M, Randall R, Chowdhury HM, Snape KM, Bradshaw TY, Southgate L, Lee GJ, Jackson I, Lord GM, Gibbs JS, Wilkins MR, Ohta-Ogo K, Nakamura K, Girerd B, Coulet F, Soubrier F, Humbert M, Morrell NW, Trembath RC, Machado RD. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat. 2011;32:1385-9. [PubMed: 21898662]
- Oldenburg MS, Frisch CD, Lindor NM, Edell ES, Kasperbauer JL, O'Brien EK. Myhre-LAPs syndrome and intubation related airway stenosis: keys to diagnosis and critical therapeutic interventions. Am J Otolaryngol. 2015;36:636-41. [PubMed: 25940662]
- Picco P, Naselli A, Pala G, Marsciani A, Buoncompagni A, Martini A. Recurrent pericarditis in Myhre syndrome. Am J Med Genet Part A 161A. 2013;1164-6. [PubMed: 23610053]
- Piccolo P, Mithbaokar P, Sabatino V, Tolmie J, Melis D, Schiaffino MC, Filocamo M, Andria G, Brunetti-Pierri N. SMAD4 mutations causing Myhre syndrome result in disorganization of extracellular matrix improved by losartan. Eur J Hum Genet. 2014;22:988-94. [PMC free article: PMC3984901] [PubMed: 24398790]
- Rahbari R, Wuster A, Lindsay SJ, Hardwick RJ, Alexandrov LB, Turki SA, Dominiczak A, Morris A, Porteous D, Smith B, Stratton MR, Hurles ME, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126-33. [PMC free article: PMC4731925] [PubMed: 26656846]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Starr LJ, Grange DK, Delaney JW, Yetman AT, Hammel JM, Sanmann JN, Perry DA, Schaefer GB, Olney AH. Myhre syndrome: Clinical features and restrictive cardiopulmonary complications. Am J Med Genet A. 2015;167A:2893-901. [PubMed: 26420300]
- Starr LJ, Lindsay ME, Perry D, Gheewalla G, VanderLaan PA, Majid A, Strange C, Costea GC, Lungu A, Lin AE. Review of the pathologic characteristics in Myhre syndrome: gain-of-function pathogenic variants in SMAD4 cause a multisystem fibroproliferative response. Pediatr Dev Pathol. 2022;25:611-23. [PubMed: 36120950]
- Yang DD, Rio M, Michot C, Boddaert N, Yacoub W, Garcelon N, Thierry B, Bonnet D, Rondeau S, Herve D, Guey S, Angoulvant F, Cormier-Daire V. Natural history of Myhre syndrome. Orphanet J Rare Dis. 2022;17:304. [PMC free article: PMC9338657] [PubMed: 35907855]
Publication Details
Author Information and Affiliations
Boston, Massachusetts
University of Naples "Federico II"
Naples, Italy
Boston, Massachusetts
Rochester, Minnesota
Omaha, Nebraska
Publication History
Initial Posting: April 13, 2017; Last Update: November 24, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Lin AE, Brunetti-Pierri N, Lindsay ME, et al. Myhre Syndrome. 2017 Apr 13 [Updated 2022 Nov 24]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.






