Summary
Clinical characteristics.
Cranioectodermal dysplasia (CED) is a ciliopathy with skeletal involvement (narrow thorax, shortened proximal limbs, syndactyly, polydactyly, brachydactyly), ectodermal features (widely spaced hypoplastic teeth, hypodontia, sparse hair, skin laxity, abnormal nails), joint laxity, growth deficiency, and characteristic facial features (frontal bossing, low-set simple ears, high forehead, telecanthus, epicanthal folds, full cheeks, everted lower lip). Most affected children develop nephronophthisis that often leads to end-stage kidney disease in infancy or childhood, a major cause of morbidity and mortality. Hepatic fibrosis and retinal dystrophy are also observed. Dolichocephaly, often secondary to sagittal craniosynostosis, is a primary manifestation that distinguishes CED from most other ciliopathies. Brain malformations and developmental delay may also occur.
Diagnosis/testing.
The diagnosis of CED is established in a proband with characteristic clinical and radiographic features (including two frequent features and two other abnormalities, with at least one ectodermal defect – i.e., involvement of the teeth, hair, or nails) and/or by identification of biallelic pathogenic variants in one of the six genes currently known to be associated with CED: IFT43, IFT52, IFT122, IFT140, WDR19, or WDR35.
Management.
Treatment of manifestations: As needed, surgery to correct sagittal craniosynostosis usually before age one year. Surgical correction may be needed for polydactyly of the hands and feet. Orthopedic care for hip dysplasia. Human growth hormone therapy could be considered in those who meet standard treatment criteria. Standard treatment for dental anomalies, nephronophthisis, liver disease, cardiac anomalies, and/or inguinal and umbilical hernias. For those with progressive visual impairment: low-vision aids and appropriate educational programs. Mechanical ventilation may be required in newborns with pulmonary hypoplasia. For those with developmental delay, speech and physical therapy and appropriate educational programs.
Surveillance: In infancy and childhood: monitor tooth development; morning urine osmolarity testing, urine collection assay for polyuria, blood pressure, serum creatinine and blood urea assessment, and renal ultrasound as recommended by nephrologist; hepatic transaminases and measurement of synthetic liver function as recommended by hepatologist. Annual ophthalmologic examinations starting at age four years to detect early signs of retinal degeneration. Cardiac examinations, EKG, and echocardiography per cardiologist. Review of developmental progress at each primary care visit, and formal evaluation with neuropsychological testing if delays noted.
Genetic counseling.
CED is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a CED-causing pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Carrier testing for at-risk relatives, prenatal testing for a pregnancy at increased risk, and preimplantation genetic testing are possible once the CED-causing pathogenic variants have been identified in an affected family member. Second-trimester ultrasound examination may detect renal cysts, shortening of the limbs, and/or polydactyly.
Diagnosis
Suggestive Findings
Cranioectodermal dysplasia (CED) should be suspected in individuals with the following clinical and radiographic findings.
Frequent features (in >75%)
- Characteristic facial features (e.g., frontal bossing, low-set/simple ears, high forehead, telecanthus, epicanthal folds, full cheeks, everted lower lip)
- Brachydactyly
- Dolichocephaly and sagittal craniosynostosis
- Shortening (and bowing) of proximal bones (mostly humeri)
- Short stature
Common features (50%-75%)
- Narrow thorax (with dysplastic ribs and pectus excavatum)
- Dental abnormalities (malformed, widely spaced teeth, and/or hypodontia)
- Sparse and/or thin hair
- Nephronophthisis (a phenotype of progressive kidney disease that may include features such as renal cysts, scarring, echogenic kidneys on ultrasound, chronic tubulointerstitial nephritis, and reduced renal function / renal concentrating ability)
- Developmental delay (most often affecting motor development)
Less common features (25%-50%)
- Joint laxity
- Liver disease (hepatic fibrosis, cirrhosis, and/or hepatomegaly)
- Syndactyly
- Polydactyly
- Abnormal nails
- Skin laxity
- Recurrent lung infections
- Bilateral inguinal hernias
Occasional to infrequent features (<25%)
- Retinal dystrophy
- Hip dysplasia
- Cystic hygroma
- Congenital heart defect
- Intellectual disability
Note: Some suggestive findings (e.g., developmental delay, dental abnormalities, abnormalities of the retina, kidney, and liver) may not be present in a neonate at the time of evaluation.
Establishing the Diagnosis
Although formal evidence-based diagnostic criteria have not been delineated, the clinical diagnosis of CED can be established in a proband based on clinical diagnostic criteria [Lin et al 2013], or the molecular diagnosis can be established in a proband with suggestive findings and biallelic pathogenic (or likely pathogenic) variants in one of the genes listed in Table 1.
Note: (1) Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include any likely pathogenic variants. (2) Identification of biallelic variants of uncertain significance (or of one known pathogenic variant and one variant of uncertain significance) in one of the genes listed in Table 1 does not establish or rule out the diagnosis.
Clinical diagnosis. The clinical diagnosis of CED can be established in a proband with characteristic clinical and radiographic features described in Suggestive Findings including two frequent features and two other abnormalities, at least one of which is an ectodermal defect (i.e., involvement of the teeth, hair, or nails). Sagittal craniosynostosis distinguishes CED from most other ciliopathies [Lin et al 2013].
Molecular diagnosis. The molecular diagnosis of CED is established in a proband with Suggestive Findings and biallelic pathogenic variants in one of the genes listed in Table 1 identified on molecular genetic testing.
Molecular genetic testing approaches can include a combination of gene-targeted testing (multigene panel) and comprehensive genomic testing (exome sequencing, genome sequencing) depending on the phenotype.
Gene-targeted testing requires that the clinician determine which gene(s) are likely involved, whereas genomic testing does not. Individuals with the distinctive findings described in Suggestive Findings are likely to be diagnosed using gene-targeted testing (see Option 1), whereas those with a phenotype indistinguishable from many other inherited disorders with ectodermal dysplasia and/or skeletal anomalies are more likely to be diagnosed using genomic testing (see Option 2).
Option 1
A multigene panel that includes the genes listed in Table 1 and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
When the phenotype is indistinguishable from many other inherited disorders characterized by ectodermal dysplasia and/or skeletal anomalies, comprehensive genomic testing, which does not require the clinician to determine which gene(s) are likely involved, is the best option. Exome sequencing is most commonly used; genome sequencing is also possible.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in Cranioectodermal Dysplasia
Clinical Characteristics
Clinical Description
Cranioectodermal dysplasia (CED) is a ciliopathy with significant involvement of the skeleton, ectoderm (teeth, hair, and nails), retina, kidneys, liver, lungs, and occasionally the brain. The current understanding of the CED phenotype is limited by the small number of well-described affected individuals reported and the even smaller number with a molecularly confirmed diagnosis.
To date, 44 individuals with biallelic pathogenic variants in one of the genes listed in Table 1 have been described clinically [Zaffanello et al 2006; Fry et al 2009; Gilissen et al 2010; Walczak-Sztulpa et al 2010; Arts et al 2011; Bredrup et al 2011; Bacino et al 2012; Hoffer et al 2013; Lin et al 2013; Alazami et al 2014; Tsurusaki et al 2014; Li et al 2015; Daoud et al 2016; Girisha et al 2016; Moosa et al 2016; Smith et al 2016; Antony et al 2017; Bayat et al 2017; Silveira et al 2017; Walczak-Sztulpa et al 2017; Xu et al 2017; Yoshikawa et al 2017; Ackah et al 2018; Córdova-Fletes et al 2018; Walczak-Sztulpa et al 2018; Walczak-Sztulpa et al 2020; Author, personal communication]. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Select Features of Cranioectodermal Dysplasia
Characteristic facial features can be observed from birth and are evident in nearly all individuals with CED (see Figure 1), including frontal bossing, bitemporal narrowing, and a tall forehead; low-set, simple, and/or posteriorly rotated ears; telecanthus, epicanthal folds, and/or down-/upslanting palpebral fissures; full cheeks; micrognathia; everted lower lip; and anteverted nares.
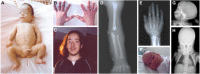
Figure 1.
Clinical and radiographic features of cranioectodermal dysplasia (CED) Patient 1 (A-E):
Dolichocephaly (apparently increased anteroposterior length of the head compared to width) and frontal bossing are usually secondary to sagittal craniosynostosis, which is usually present at birth. Sib pairs may show discordance for sagittal craniosynostosis [Arts et al 2011, Bredrup et al 2011].
Skeletal findings
- Hands and feet. Prenatal ultrasound may detect polydactyly during mid-gestation [Konstantinidou et al 2009]. Neonates often have brachydactyly (with short middle and distal phalanges that may be abnormally shaped), postaxial polydactyly, and cutaneous syndactyly of fingers and toes (most frequently mild cutaneous syndactyly of second and third toes). Epiphyses of phalanges can have a normal appearance on radiograph or can be flattened or cone shaped [Fry et al 2009]. Other findings of the hands and feet variably seen include fifth-finger clinodactyly, abnormal palmar creases, restricted finger flexion, osteoporosis, sandal gap, and/or triphalangeal hallux [Gilissen et al 2010, Bacino et al 2012, Hoffer et al 2013, Lin et al 2013].
- Thorax. A narrow thorax with short dysplastic ribs is common, though not ubiquitous, and may be noted as early as mid-gestation; however, this finding was most commonly first noted at birth [Lin et al 2013]. Pectus excavatum is often observed [Hoffer et al 2013]. Rib deformities (e.g., short ribs, coat-hanger-shaped ribs) may normalize during childhood [Bacino et al 2012].
- Shortening (and bowing) of proximal long bones has been noted as early as 23 weeks' gestation. Upper limbs are often shorter compared to lower limbs; humeri are particularly affected. Long bones may display bowing, and epiphyses may be flattened and/or display metaphyseal flaring [Bacino et al 2012, Hoffer et al 2013, Lin et al 2013].
- Growth deficiency. At birth the length as related to gestational age may be within the normal range but can also be below the third centile. Infants may have growth deficiency with length below the third centile, but length may also be between the fifth and tenth centile [Bacino et al 2012].
Ectodermal defects
- Teeth. Tooth eruption is often delayed. Deciduous teeth are generally small and widely spaced. Hypodontia, enamel defects, taurodontia, and fused and cone-shaped teeth have also been reported. Similar characteristics are seen in permanent teeth. Hypo- or oligodontia may affect upper as well as lower permanent teeth [Fry et al 2009, Gilissen et al 2010, Walczak-Sztulpa et al 2010, Arts et al 2011, Bredrup et al 2011].
- Skin. Prenatal ultrasound may reveal mid-gestational nuchal webbing and skin thickening. Generalized skin laxity and redundant skin folds have been reported in infancy and thereafter. Skin folds have been found particularly at the neck, ankles, and wrists. Skin may be dry; hyperkeratosis has been reported [Arts et al 2011, Bredrup et al 2011, Bacino et al 2012].
- Hair. Most infants and young children with CED have sparse, fine hair. Hair may be hypopigmented with reduced diameter. Hair growth may also be affected [Arts et al 2011, Lin et al 2013]. In some instances, hair growth may normalize during childhood [Konstantinidou et al 2009].
- Nails are short, broad, and slow growing from infancy [Hoffer et al 2013].
Kidney disease is due to nephronophthisis (tubulointerstitial nephritis). At least 60%-70% of persons with CED were reported to have renal insufficiency. Although end-stage kidney disease (ESKD) can be evident prenatally as poly/oligohydramnios and small cystic kidneys in the second trimester of pregnancy, the first signs of kidney disease are often evident in early childhood (age ~2 years) [Bacino et al 2012]. Initially reduced urinary concentrating ability leads to polyuria and polydipsia. Nocturnal enuresis may be evident. Hypertension, proteinuria, hematuria, and electrolyte imbalances usually develop later in the disease course as a result of renal insufficiency and filtration defects. In ten of 21 children kidney disease progressed to ESKD. Of note, this number may have increased over time as follow-up studies are limited. Most children developed ESKD between ages two and six years [Hoffer et al 2013, Lin et al 2013].
Renal ultrasound examination in infancy and early childhood usually shows normal-sized or small kidneys with increased echogenicity and poor corticomedullary differentiation [Lin et al 2013]. Renal biopsy shows interstitial fibrosis with focal inflammatory cell infiltrates, tubular atrophy, glomerulosclerosis, and occasional cysts [Obikane et al 2006, Konstantinidou et al 2009, Bredrup et al 2011, Lin et al 2013]. The latter features occur in advanced disease.
Liver findings range from hepatosplenomegaly without signs of progressive liver disease to extensive liver abnormalities including (recurrent) hyperbilirubinemia and cholestatic disease requiring hospitalization in the newborn period [Walczak-Sztulpa et al 2010, Bacino et al 2012]. Liver cirrhosis, severe cholestasis with bile duct proliferation, and acute cholangitis have been described in infants [Zaffanello et al 2006, Arts et al 2011, Bacino et al 2012, Lin et al 2013]. Liver cysts have been detected in children age three and four years [Hoffer et al 2013], but also as early as age ten months [Lin et al 2013]. Longitudinal data on liver disease are not available; however, the long-term prognosis with respect to liver fibrosis and cirrhosis is probably poor.
Eye findings include retinal dystrophy and nystagmus [Bredrup et al 2011, Lin et al 2013]. Nyctalopia (night blindness) is often evident in the first years of life [Bredrup et al 2011]. Abnormal scotopic and photopic electroretinograms have been reported as early as ages four to 11 years, while fundoscopy has revealed attenuated arteries and bone-spicule-shaped deposits as early as ages five to 11 years in some [Bredrup et al 2011]. The natural history of the retinal dystrophy remains to be reported; however, in overlapping ciliopathies such as Bardet-Biedl syndrome, night blindness usually progresses to legal blindness in young adults (see Bardet-Biedl Syndrome). A similar prognosis is to be expected in CED.
Other ophthalmologic findings include hyperopia, myopia, esotropia, myopic/hypermetropic astigmatism, and euryblepharon (excess horizontal eyelid length) [Konstantinidou et al 2009, Bredrup et al 2011, Hoffer et al 2013, Lin et al 2013].
Pulmonary. In infancy or early childhood, children with CED may experience life-threatening respiratory distress due to pulmonary hypoplasia and recurrent respiratory infections. Asthma and pneumothorax have also been reported [Bredrup et al 2011, Bacino et al 2012, Hoffer et al 2013]. Many children die of respiratory distress after birth or of pneumonia during early childhood [Tamai et al 2002]. Recurrent respiratory infections have been reported to diminish in frequency over time [Konstantinidou et al 2009].
Cardiac malformations have included patent ductus arteriosus and atrial and ventricular septal defects. Thickening of the mitral and tricuspid valves, ventricular hypertrophy/dilatation, and peripheral pulmonary stenosis have also been reported [Arts et al 2011, Bacino et al 2012]. Bacino et al [2012] reported one affected child in whom cardiac arrhythmia and atrial septal defect resolved at age three years.
Central nervous system. Although the majority of children develop normally, mild developmental delay is reported in some individuals [Hoffer et al 2013, Lin et al 2013]. Sitting unsupported may be delayed to nine to 15 months and walking to three years [Fry et al 2009, Bacino et al 2012, Hoffer et al 2013]. Delays in speech may vary from a few words at age 19 months to no words at age five years [Hoffer et al 2013]. No information is available on how affected individuals respond to speech and physical therapy. Cognitive and social abilities are usually normal but intellectual disability is diagnosed in some individuals [Fry et al 2009, Alazami et al 2014, Li et al 2015].
Brain imaging has revealed cortical atrophy, ventriculomegaly, large cisterna magna, hypoplasia of the corpus callosum, focal microdysgenesis, enlarged extracerebral fluid spaces, large posterior fossa cyst, and Dandy-Walker malformation [Zannolli et al 2001, Fry et al 2009, Konstantinidou et al 2009, Bacino et al 2012, Hoffer et al 2013, Lin et al 2013, Girisha et al 2016, Walczak-Sztulpa et al 2020].
Other
- Joint laxity can be observed from the neonatal period [Fry et al 2009].
- Hernia. (Bilateral) inguinal hernias and/or umbilical hernia can be present in neonates or during the first year of life [Fry et al 2009, Walczak-Sztulpa et al 2010].
Life expectancy. Morbidity is high in individuals with CED and hospitalization may be frequent and/or extended [Bacino et al 2012]. Mortality rates are unclear, although 10/65 children with CED died before age seven years of respiratory failure [Levin et al 1977, Savill et al 1997, Tamai et al 2002], heart failure [Eke et al 1996, Savill et al 1997, Bacino et al 2012, Silveira et al 2017, Walczak-Sztulpa et al 2017], hypovolemic shock (as a result of coagulopathy) [Bacino et al 2012], or unknown causes [Lin et al 2013, Antony et al 2017]. This number could be higher as longitudinal data on the majority of individuals with CED are unavailable. At least three persons with CED survived into young adulthood (see Bredrup et al [2011] and Figure 1).
Phenotype Correlations by Gene
Phenotypes resulting from biallelic pathogenic variants in any one of the six known genes (i.e., IFT43, IFT52, IFT122, IFT140, WDR19, WDR35) are not distinguishable [Walczak-Sztulpa et al 2010, Arts et al 2011, Bredrup et al 2011, Bacino et al 2012, Hoffer et al 2013, Girisha et al 2016, Bayat et al 2017].
Genotype-Phenotype Correlations
No genotype-phenotype correlations have been confirmed.
Nomenclature
CED was first described as Sensenbrenner syndrome in a sib pair with dolichocephaly, rhizomelic shortening of the bones, brachydactyly, and ectodermal defects [Sensenbrenner et al 1975]. Subsequently Levin et al [1977] described affected individuals from two additional families and renamed the disorder cranioectodermal dysplasia.
Prevalence
CED is rare; its exact frequency is unknown. Fewer than 100 affected individuals have been reported.
Genetically Related (Allelic) Disorders
No phenotypes other than those discussed in this GeneReview are known to be associated with pathogenic variants in IFT122. Note: An individual with biallelic IFT122 pathogenic variants was diagnosed with Beemer-Langer syndrome (short-rib polydactyly syndrome, type IV); on further review, however, the phenotype in this individual is more consistent with cranioectodermal dysplasia (CED) [Silveira et al 2017].
Heterozygous pathogenic variants in IFT140 have been identified in individuals with a mild polycystic kidney disease phenotype (see Polycystic Kidney Disease, Autosomal Dominant).
Biallelic pathogenic variants in IFT43 and IFT140 have been associated with isolated retinitis pigmentosa 81 (OMIM 617871) and isolated retinitis pigmentosa 80 (OMIM 617781), respectively. Other autosomal recessive phenotypes associated with germline pathogenic variants in IFT43, IFT52, IFT140, WDR19, and WDR35 are summarized in Table 3. These disorders have phenotypic features that overlap with CED and should be considered in the Differential Diagnosis.
Table 3.
Allelic Disorders to Consider in the Differential Diagnosis of Cranioectodermal Dysplasia
Differential Diagnosis
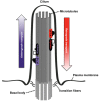
Cranioectodermal dysplasia (CED) is part of a spectrum of disorders caused by disruption of the cilium, an organelle of the cell that appears and functions as an antenna (see Figure 2) [Huber & Cormier-Daire 2012]. These disorders, collectively referred to as ciliopathies, display marked phenotypic overlap. Typical clinical features of ciliopathies are: renal cystic disease; retinal dystrophy; shortening of ribs, phalanges, and long bones; polydactyly; hepatic fibrosis; and developmental delay.
Within the ciliopathies, Jeune asphyxiating thoracic dystrophy, Mainzer-Saldino syndrome, Ellis-van Creveld syndrome, and the short-rib polydactyly syndromes resemble CED the most (see Table 4). These autosomal recessive skeletal ciliopathies are referred to as short-rib thoracic dysplasia in OMIM (OMIM PS208500).
Table 4.
Skeletal Ciliopathies of Interest in the Differential Diagnosis of Cranioectodermal Dysplasia
Other ciliopathies that clinically overlap with CED include isolated nephronophthisis, isolated retinal dystrophy, Caroli disease, Senior-Løken syndrome, Joubert syndrome, Meckel-Gruber syndrome (OMIM PS249000), and Bardet-Biedl syndrome [Huber & Cormier-Daire 2012].
- Senior-Løken syndrome (OMIM 266900) is a heterogeneous autosomal recessive disorder that is characterized by nephronophthisis and retinal dystrophy. Pathogenic variants in CEP290, IQCB1, NPHP1, NPHP4, SDCCAG8, TRAF3IP1, and WDR19 have been detected in persons with Senior-Løken syndrome. (Note: Pathogenic variants in WDR19 are also associated with CED.)
- Caroli disease (OMIM 600643) is characterized by polycystic liver disease and cholangitis. It is part of the autosomal recessive polycystic kidney disease spectrum of disorders and can occur as an isolated finding as well as in combination with other features including renal cystic disease.
- Autosomal recessive retinal dystrophy (also known as retinitis pigmentosa) can be an isolated finding or occur in syndromic disorders such as CED. Retinitis pigmentosa usually starts with night blindness and can progress to complete blindness later in life due to loss of the photoreceptors (rods and cones). The fundus often displays attenuation of retinal vessels and may reveal abnormal peripheral pigmentation (referred to as bone-spicule deposits). More than 50% of families with isolated retinitis pigmentosa have an autosomal recessive form. Pathogenic variants in more than 50 genes can cause RP, and almost two thirds of these genes encode ciliary proteins.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of a newborn or infant diagnosed with cranioectodermal dysplasia (CED), the evaluations summarized in Table 5 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 5.
Recommended Evaluations Following Initial Diagnosis in Individuals with Cranioectodermal Dysplasia
Treatment of Manifestations
Table 6.
Treatment of Manifestations in Individuals with Cranioectodermal Dysplasia
Surveillance
Table 7.
Recommended Surveillance for Individuals with Cranioectodermal Dysplasia
Agents/Circumstances to Avoid
If kidney disease is present, a nephrologist may recommend reduction of potassium and phosphorus from the diet.
Nephrotoxic medications, including the NSAID class of drugs, may also be a relative contraindication in individuals with kidney involvement. Individuals should be under the care of a nephrologist, if indicated, to discuss what nephrotoxic agents to avoid.
Evaluation of Relatives at Risk
If the CED-causing pathogenic variants have been identified in an affected family member, it is appropriate to clarify the genetic status of at-risk infants to allow early diagnosis and appropriate management and surveillance, particularly for respiratory complications, renal and liver disease, and visual impairment.
If the pathogenic variants are not known in a family, the following is recommended for at-risk children:
- In the newborn. Physical examination by a pediatrician; consultation with a clinical geneticist as determined by the clinical findings
- By age three months. Kidney and liver evaluation including ultrasound examination and measurement of blood pressure, serum creatinine concentration, and liver enzymes
- At six-month intervals. Repeat kidney and liver evaluation
Parents should be alerted to the signs of CED and advised to contact their child's health care provider if suspicious symptoms, such as polydipsia and/or jaundice, appear.
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
Cranioectodermal dysplasia (CED) is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected child are obligate heterozygotes (i.e., presumed to be carriers of one CED-causing pathogenic variant based on family history).
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for a CED-causing pathogenic variant and to allow reliable recurrence risk assessment. If a pathogenic variant is detected in only one parent, the following possibilities should be considered:
- One of the pathogenic variants identified in the proband occurred as a de novo event in the proband or as a postzygotic de novo event in a mosaic parent [Walczak-Sztulpa et al 2010, Jónsson et al 2017].
- Uniparental isodisomy for the parental chromosome with the pathogenic variant resulted in homozygosity for the pathogenic variant in the proband.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- If both parents are known to be heterozygous for a CED-causing pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Clinical manifestations of CED are highly variable and may differ among affected sibs.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. Due to the rarity of CED and lack of longitudinal follow up, it is currently unknown whether individuals with CED are fertile. No individuals with CED have been reported to reproduce. This may be due to the severity of the disorder and to limited life expectancy.
Other family members. If both parents are known to be heterozygous for a CED-causing pathogenic variant, each sib of the proband's parents is at a 50% risk of being a carrier.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the CED-causing pathogenic variants in the family.
Related Genetic Counseling Issues
See Management, Evaluation of Relatives at Risk for information on evaluating at-risk relatives for the purpose of early diagnosis and treatment.
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
DNA banking. Because it is likely that testing methodology and our understanding of genes, pathogenic mechanisms, and diseases will improve in the future, consideration should be given to banking DNA from probands in whom a molecular diagnosis has not been confirmed (i.e., the causative pathogenic mechanism is unknown). For more information, see Huang et al [2022].
Prenatal Testing and Preimplantation Genetic Testing
Molecular genetic testing. Once the CED-causing pathogenic variants have been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Fetal ultrasound examination. Second-trimester ultrasound examination may detect renal cysts, shortening of the limbs, and/or polydactyly.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- National Foundation for Ectodermal Dysplasias (NFED)Phone: 618-566-2020Email: info@nfed.org
- Ectodermal Dysplasias International RegistryEmail: info@nfed.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Cranioectodermal Dysplasia: Genes and Databases
Table B.
OMIM Entries for Cranioectodermal Dysplasia (View All in OMIM)
Molecular Pathogenesis
Cranioectodermal dysplasia (CED) belongs to a spectrum of disorders known as ciliopathies [Baker & Beales 2009, Konstantinidou et al 2009, Arts & Knoers 2013]. Ciliopathies are thought to result from defects in cilia (see Figure 2), projections from the cell that occur almost ubiquitously throughout the human body. These microtubule-based organelles are thought to function as signaling hubs regulating pathways that are essential for normal human development and tissue homeostasis [Hildebrandt et al 2011].
A process that is required for ciliogenesis and regulation of signaling pathways is ciliary transport (also known as intraflagellar transport [IFT]) [Hildebrandt et al 2011, Taschner et al 2012]. Upward (anterograde) movement of cargo or signaling molecules occurs through the multi-subunit IFT-B complex in association with the heterotrimeric kinesin-2 motor, while downward (i.e., tip-to-base [retrograde]) ciliary transport is regulated by the dynein-2 motor in association with the IFT-A complex [Hildebrandt et al 2011, Taschner et al 2012].
Most pathogenic variants in individuals with CED identified to date occur in genes that encode members of the IFT-A hexamere protein complex: IFT122 (previously WDR10), WDR35 (IFT121), WDR19 (IFT144), IFT43 (previously C14orf179), or IFT140 [Gilissen et al 2010, Arts et al 2011, Bredrup et al 2011, Bacino et al 2012, Hoffer et al 2013, Lin et al 2013, Caparrós-Martín et al 2015]. The remaining IFT-A complex member is encoded by TTC21B. Of note, genes encoding the proteins IFT139, IFT140, and DYNC2H1 (a subunit of dynein-2 motor) are mutated in disorders that clinically overlap with CED (see Differential Diagnosis) [Dagoneau et al 2009, Davis et al 2011, Perrault et al 2012].
When the IFT-A protein complex is disrupted in CED, cilia in fibroblasts have bulging tips, which contain accumulations of IFT-B complex proteins that normally regulate base-to-tip (anterograde) ciliary transport [Arts et al 2011, Bredrup et al 2011].
Pathogenic variants in IFT52, which encodes part of the IFT-B protein complex, have also been reported to cause CED [Girisha et al 2016].
Although shortening of cilia in fibroblasts from persons with CED has also been reported [Walczak-Sztulpa et al 2010], this is not always observed [Bredrup et al 2011]. It is thought that defective IFT and resulting structural defects in the ciliary architecture disturb important developmental signaling cascades (e.g., hedgehog signaling), resulting in CED [Walczak-Sztulpa et al 2010, Ocbina et al 2011, Qin et al 2011, Liem et al 2012].
Mechanism of disease causation. Loss of function
Gene-specific laboratory considerations. A pathogenic variant in IFT140 resulting in a tandem duplication has been identified.
Chapter Notes
Author Notes
This work was funded by the grants from the Dutch Kidney Foundation (CP11.18 "KOUNCIL" to N Knoers and H Arts) and the Netherlands Organization for Health Research and Development (ZonMw 91613008 to H Arts); both projects aim to unravel the genetics and mechanisms of disease of renal ciliopathies including Sensenbrenner syndrome.
Acknowledgments
We are thankful for the participation of the Sensenbrenner syndrome families in our study and for their consent for publication of clinical images. We also thank Dr E Lapi and Dr E Andreucci for their clinical contribution, Dr C Marcelis, Dr N van de Kar, Dr L Koster-Kamphuis, and Dr K Noordam for useful discussions, and Prof Dr HG Brunner for communication.
Author History
Heleen Arts, PhD; McMaster University (2013-2021)
Kim Keppler-Noreuil, MD (2021-present)
Nine Knoers, MD, PhD; University Medical Center Groningen (2013-2021)
Angela Lin, MD (2021-present)
Weizhen Tan, MD (2021-present)
Revision History
- 15 December 2022 (aa) Revision: added IFT140-related autosomal dominant polycystic kidney disease to Genetically Related Disorders
- 11 March 2021 (sw) Comprehensive update posted live
- 12 April 2018 (ha) Revision: Lin et al [2013] added
- 12 September 2013 (me) Review posted live
- 26 April 2012 (ha) Original submission
References
Literature Cited
- Ackah RL, Yoeli D, Kueht M, Galván NTN, Cotton RT, Rana A, O'Mahony CA, Goss JA. Orthotopic liver transplantation for Sensenbrenner syndrome. Pediatr Transplant. 2018:22. [PubMed: 29076289]
- Alazami AM, Seidahmed MZ, Alzahrani F, Mohammed AO, Alkuraya FS. Novel IFT122 mutation associated with impaired ciliogenesis and cranioectodermal dysplasia. Mol Genet Genomic Med. 2014;2:103–6. [PMC free article: PMC3960051] [PubMed: 24689072]
- Antony D, Nampoory N, Bacchelli C, Melhem M, Wu K, James CT, Beales PL, Hubank M, Thomas D, Mashankar A, Behbehani K, Schmidts M, Alsmadi O. Exome sequencing for the differential diagnosis of ciliary chondrodysplasias: Example of a WDR35 mutation case and review of the literature. Eur J Med Genet. 2017;60:658–66. [PubMed: 28870638]
- Arts HH, Bongers EM, Mans DA, van Beersum SE, Oud MM, Bolat E, Spruijt L, Cornelissen EA, Schuurs-Hoeijmakers JH, de Leeuw N, Cormier-Daire V, Brunner HG, Knoers NV, Roepman R. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J Med Genet. 2011;48:390–5. [PubMed: 21378380]
- Arts HH, Knoers NV. Current insights into renal ciliopathies: what can genetics teach us? Pediatr Nephrol. 2013;28:863–74. [PMC free article: PMC3631122] [PubMed: 22829176]
- Bacino CA, Dhar SU, Brunetti-Pierri N, Lee B, Bonnen PE. WDR35 mutation in siblings with Sensenbrenner syndrome: a ciliopathy with variable phenotype. Am J Med Genet A. 2012;158A:2917–24. [PMC free article: PMC4000731] [PubMed: 22987818]
- Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet. 2009;151C:281–95. [PubMed: 19876933]
- Baujat G, Le Merrer M. Ellis-van Creveld syndrome. Orphanet J Rare Dis. 2007;2:27. [PMC free article: PMC1891277] [PubMed: 17547743]
- Bayat A, Kerr B, Douzgou S, et al. The evolving craniofacial phenotype of a patient with Sensenbrenner syndrome caused by IFT140 compound heterozygous mutations. Clin Dysmorphol. 2017;26:247–51. [PubMed: 28288023]
- Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, Leh SM, Midtbø M, Filhol E, Bole-Feysot C, Nitschké P, Gilissen C, Haugen OH, Sanders JS, Stolte-Dijkstra I, Mans DA, Steenbergen EJ, Hamel BC, Matignon M, Pfundt R, Jeanpierre C, Boman H, Rødahl E, Veltman JA, Knappskog PM, Knoers NV, Roepman R, Arts HH. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet. 2011;89:634–43. [PMC free article: PMC3213394] [PubMed: 22019273]
- Caparrós-Martín JA, De Luca A, Cartault F, Aglan M, Temtamy S, Otaify GA, Mehrez M, Valencia M, Vázquez L, Alessandri JL, Nevado J, Rueda-Arenas I, Heath KE, Digilio MC, Dallapiccola B, Goodship JA, Mill P, Lapunzina P, Ruiz-Perez VL. Specific variants in WDR35 cause a distinctive form of Ellis-van Creveld syndrome by disrupting the recruitment of the EvC complex and SMO into the cilium. Hum Mol Genet. 2015;24:4126–37. [PMC free article: PMC4560068] [PubMed: 25908617]
- Córdova-Fletes C, Becerra-Solano LE, Rangel-Sosa MM, Rivas-Estilla AM, Alberto Galán-Huerta K, Ortiz-López R, Rojas-Martínez A, Juárez-Vázquez CI, García-Ortiz JE. Uncommon runs of homozygosity disclose homozygous missense mutations in two ciliopathy-related genes (SPAG17 and WDR35) in a patient with multiple brain and skeletal anomalies. Eur J Med Genet. 2018;61:161–7. [PubMed: 29174089]
- Dagoneau N, Goulet M, Geneviève D, Sznajer Y, Martinovic J, Smithson S, Huber C, Baujat G, Flori E, Tecco L, Cavalcanti D, Delezoide AL, Serre V, Le Merrer M, Munnich A, Cormier-Daire V. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am J Hum Genet. 2009;84:706–11. [PMC free article: PMC2681009] [PubMed: 19442771]
- Daoud H, Luco SM, Li R, Bareke E, Beaulieu C, Jarinova O, Carson N, Nikkel SM, Graham GE, Richer J, Armour C, Bulman DE, Chakraborty P, Geraghty M, Lines MA, Lacaze-Masmonteil T, Majewski J, Boycott KM, Dyment DA. Next-generation sequencing for diagnosis of rare diseases in the neonatal intensive care unit. CMAJ. 2016;188:E254–E260. [PMC free article: PMC4978597] [PubMed: 27241786]
- Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, Stoetzel C, Szymanska K, Ramaswami G, Logan CV, Muzny DM, Young AC, Wheeler DA, Cruz P, Morgan M, Lewis LR, Cherukuri P, Maskeri B, Hansen NF, Mullikin JC, Blakesley RW, Bouffard GG, Gyapay G, Rieger S, Tönshoff B, Kern I, Soliman NA, Neuhaus TJ, Swoboda KJ, Kayserili H, Gallagher TE, Lewis RA, Bergmann C, Otto EA, Saunier S, Scambler PJ, Beales PL, Gleeson JG, Maher ER, Attié-Bitach T, Dollfus H, Johnson CA, Green ED, Gibbs RA, Hildebrandt F, Pierce EA, Katsanis N, et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011;43:189–96. [PMC free article: PMC3071301] [PubMed: 21258341]
- Eke T, Woodruff G, Young ID. A new oculorenal syndrome: retinal dystrophy and tubulointerstitial nephropathy in cranioectodermal dysplasia. Br J Ophthalmol. 1996;80:490–1. [PMC free article: PMC505510] [PubMed: 8695580]
- Elçioglu NH, Hall CM. Diagnostic dilemmas in the short rib-polydactyly syndrome group. Am J Med Genet. 2002;111:392–400. [PubMed: 12210298]
- Fry AE, Klingenberg C, Matthes J, Heimdal K, Hennekam RC, Pilz DT. Connective tissue involvement in two patients with features of cranioectodermal dysplasia. Am J Med Genet A. 2009;149A:2212–5. [PubMed: 19760620]
- Gilissen C, Arts HH, Hoischen A, Spruijt L, Mans DA, Arts P, van Lier B, Steehouwer M, van Reeuwijk J, Kant SG, Roepman R, Knoers NV, Veltman JA, Brunner HG. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet. 2010;87:418–23. [PMC free article: PMC2933349] [PubMed: 20817137]
- Girisha KM, Shukla A, Trujillano D, Bhavani GS, Hebbar M, Kadavigere R, Rolfs A. A homozygous nonsense variant in IFT52 is associated with a human skeletal ciliopathy. Clin Genet. 2016;90:536–9. [PubMed: 26880018]
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–43. [PMC free article: PMC3640822] [PubMed: 21506742]
- Hoffer JL, Fryssira H, Konstantinidou AE, Ropers HH, Tzschach A. Novel WDR35 mutations in patients with cranioectodermal dysplasia (Sensenbrenner syndrome). Clin Genet. 2013;83:92–5. [PubMed: 22486404]
- Huang SJ, Amendola LM, Sternen DL. Variation among DNA banking consent forms: points for clinicians to bank on. J Community Genet. 2022;13:389–97. [PMC free article: PMC9314484] [PubMed: 35834113]
- Huber C, Cormier-Daire V. Ciliary disorder of the skeleton. Am J Med Genet C Semin Med Genet. 2012;160C:165–74. [PubMed: 22791528]
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Konstantinidou AE, Fryssira H, Sifakis S, Karadimas C, Kaminopetros P, Agrogiannis G, Velonis S, Nikkels PG, Patsouris E. Cranioectodermal dysplasia: a probable ciliopathy. Am J Med Genet A. 2009;149A:2206–11. [PubMed: 19760621]
- Levin LS, Perrin JC, Ose L, Dorst JP, Miller JD, McKusick VA. A heritable syndrome of craniosynostosis, short thin hair, dental abnormalities, and short limbs: cranioectodermal dysplasia. J Pediatr. 1977;90:55–61. [PubMed: 830894]
- Li Y, Garrod AS, Madan-Khetarpal S, Sreedher G, McGuire M, Yagi H, Klena NT, Gabriel GC, Khalifa O, Zahid M, Panigrahy A, Weiner DJ, Lo CW. Respiratory motile cilia dysfunction in a patient with cranioectodermal dysplasia. Am J Med Genet A. 2015;167A:2188–96. [PubMed: 25914204]
- Liem KF Jr, Ashe A, He M, Satir P, Moran J, Beier D, Wicking C, Anderson KV. The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J Cell Biol. 2012;197:789–800. [PMC free article: PMC3373400] [PubMed: 22689656]
- Lin AE, Traum AZ, Sahai I, Keppler-Noreuil K, Kukolich MK, Adam MP, Westra SJ, Arts HH. Sensenbrenner syndrome (cranioectodermal dysplasia): clinical and molecular analyses of 39 patients including two new patients. Am J Med Genet A. 2013;161A:2762–76. [PubMed: 24123776]
- Moosa S, Obregon MG, Altmüller J, Thiele H, Nürnberg P, Fano V, Wollnik B. Novel IFT122 mutations in three Argentinian patients with cranioectodermal dysplasia: Expanding the mutational spectrum. Am J Med Genet A. 2016;170A:1295–301. [PubMed: 26792575]
- Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, Nishimura G, Robertson S, Sangiorgi L, Savarirayan R, Sillence D, Superti-Furga A, Unger S, Warman ML. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019;179:2393–419. [PubMed: 31633310]
- Oberklaid F, Danks DM, Mayne V, Campbell P. Asphyxiating thoracic dysplasia. Clinical, radiological, and pathological information on 10 patients. Arch Dis Child. 1977;52:758–65. [PMC free article: PMC1544803] [PubMed: 931421]
- Obikane K, Nakashima T, Watarai Y, Morita K, Cho K, Tonoki H, Nagata M, Sasaki S. Renal failure due to tubulointerstitial nephropathy in an infant with cranioectodermal dysplasia. Pediatr Nephrol. 2006;21:574–6. [PubMed: 16491415]
- Ocbina PJ, Eggenschwiler JT, Moskowitz I, Anderson KV. Complex interactions between genes controlling trafficking in primary cilia. Nat Genet. 2011;43:547–53. [PMC free article: PMC3132150] [PubMed: 21552265]
- Peña-Padilla C, Marshall CR, Walker S, Scherer SW, Tavares-Macías G, Razo-Jiménez G, Bobadilla-Morales L, Acosta-Fernández E, Corona-Rivera A, Mendoza-Londono R, Corona-Rivera JR. Compound heterozygous mutations in the IFT140 gene cause Opitz trigonocephaly C syndrome in a patient with typical features of a ciliopathy. Clin Genet. 2017;91:640–6. [PubMed: 27874174]
- Perrault I, Saunier S, Hanein S, Filhol E, Bizet AA, Collins F, Salih MA, Gerber S, Delphin N, Bigot K, Orssaud C, Silva E, Baudouin V, Oud MM, Shannon N, Le Merrer M, Roche O, Pietrement C, Goumid J, Baumann C, Bole-Feysot C, Nitschke P, Zahrate M, Beales P, Arts HH, Munnich A, Kaplan J, Antignac C, Cormier-Daire V, Rozet JM. Mainzer-Saldino syndrome is a ciliopathy caused by IFT140 mutations. Am J Hum Genet. 2012;90:864–70. [PMC free article: PMC3376548] [PubMed: 22503633]
- Qin J, Lin Y, Norman RX, Ko HW, Eggenschwiler JT. Intraflagellar transport protein 122 antagonizes Sonic Hedgehog signaling and controls ciliary localization of pathway components. Proc Natl Acad Sci U S A. 2011;108:1456–61. [PMC free article: PMC3029728] [PubMed: 21209331]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Ruiz-Perez VL, Ide SE, Strom TM, Lorenz B, Wilson D, Woods K, King L, Francomano C, Freisinger P, Spranger S, Marino B, Dallapiccola B, Wright M, Meitinger T, Polymeropoulos MH, Goodship J. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat Genet. 2000;24:283–6. [PubMed: 10700184]
- Savill GA, Young ID, Cunningham RJ, Ansell I, Evans JH. Chronic tubulo-interstitial nephropathy in children with cranioectodermal dysplasia. Pediatr Nephrol. 1997;11:215–7. [PubMed: 9090669]
- Sensenbrenner JA, Dorst JP, Owens RP. New syndrome of skeletal, dental and hair anomalies. Birth Defects Orig Artic Ser. 1975;11:372–9. [PubMed: 1227553]
- Silveira KC, Moreno CA, Cavalcanti DP. Beemer-Langer syndrome is a ciliopathy due to biallelic mutations in IFT122. Am J Med Genet A. 2017;173:1186–9. [PubMed: 28370949]
- Smith C, Lamont RE, Wade A, Bernier FP, Parboosingh JS, Innes AM. A relatively mild skeletal ciliopathy phenotype consistent with cranioectodermal dysplasia is associated with a homozygous nonsynonymous mutation in WDR35. Am J Med Genet A. 2016;170:760–5. [PubMed: 26691894]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Tamai S, Tojo M, Kamimaki T, Sato Y, Nishimura G. Intrafamilial phenotypic variations in cranioectodermal dysplasia: propositus with typical manifestations and her brother with perinatal death. Am J Med Genet. 2002;107:78–80. [PubMed: 11807876]
- Taschner M, Bhogaraju S, Lorentzen E. Architecture and function of IFT complex proteins in ciliogenesis. Differentiation. 2012;83:S12–22. [PMC free article: PMC3977345] [PubMed: 22118932]
- Tsurusaki Y, Yonezawa R, Furuya M, Nishimura G, Pooh R, Nakashima M, Saitsu H, Miyake N, Saito S, Matsumoto N. Whole exome sequencing revealed biallelic IFT122 mutations in a family with CED1 and recurrent pregnancy loss. Clin Genet. 2014;85:592–4. [PubMed: 23826986]
- Walczak-Sztulpa J, Eggenschwiler J, Osborn D, Brown DA, Emma F, Klingenberg C, Hennekam RC, Torre G, Garshasbi M, Tzschach A, Szczepanska M, Krawczynski M, Zachwieja J, Zwolinska D, Beales PL, Ropers HH, Latos-Bielenska A, Kuss AW. Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet. 2010;86:949–56. [PMC free article: PMC3032067] [PubMed: 20493458]
- Walczak-Sztulpa J, Posmyk R, Bukowska-Olech EM, Wawrocka A, Jamsheer A, Oud MM, Schmidts M, Arts HH, Latos-Bielenska A, Wasilewska A. Compound heterozygous IFT140 variants in two Polish families with Sensenbrenner syndrome and early onset end-stage renal disease. Orphanet J Rare Dis. 2020;15:36. [PMC free article: PMC6995138] [PubMed: 32007091]
- Walczak-Sztulpa J, Wawrocka A, Sobierajewicz A, Kuszel L, Zawadzki J, Grenda R, Swiader-Lesniak A, Kocyla-Karczmarewicz B, Wnuk A, Latos-Bielenska A, Chrzanowska KH. Intrafamilial phenotypic variability in a Polish family with Sensenbrenner syndrome and biallelic WDR35 mutations. Am J Med Genet A. 2017;173:1364–8. [PubMed: 28332779]
- Walczak-Sztulpa J, Wawrocka A, Swiader-Lesniak A, Socha M, Jamsheer A, Drozdz D, Latos-Bielenska A, Zachwieja K. Clinical and molecular genetic characterization of a male patient with Sensenbrenner syndrome (cranioectodermal dysplasia) and biallelic WDR35 mutations. Birth Defects Res. 2018;110:376–81. [PubMed: 29134781]
- Xu W, Jin M, Hu R, Wang H, Zhang F, Yuan S, Cao Y. The Joubert syndrome protein Inpp5e controls ciliogenesis by regulating phosphoinositides at the apical membrane. J Am Soc Nephrol. 2017;28:118–29. [PMC free article: PMC5198270] [PubMed: 27401686]
- Yoshikawa T, Kamei K, Nagata H, Saida K, Sato M, Ogura M, Ito S, Miyazaki O, Urushihara M, Kondo S, Sugawara N, Ishizuka K, Hamasaki Y, Shishido S, Morisada N, Iijima K, Nagata M, Yoshioka T, Ogata K, Ishikura K. Diversity of renal phenotypes in patients with WDR19 mutations: two case reports. Nephrology (Carlton). 2017;22:566–71. [PubMed: 28621010]
- Zaffanello M, Omedi-Camassei F, Melzi ML, Torre G, Callea F, Emma F. Sensenbrenner syndrome: a new member of the hepatorenal fibrocystic family. Am J Med Genet A. 2006;140:2336–40. [PubMed: 17022080]
- Zannolli R, Mostardini R, Carpentieri ML, Gatti MG, Galluzzi P, Terrosi Vagnoli P, Giorgetti R, Calvieri S, Morgese G. Cranioectodermal dysplasia: a new patient with an inapparent, subtle phenotype. Pediatr Dermatol. 2001;18:332–5. [PubMed: 11576410]
Publication Details
Author Information and Affiliations
Division of Pediatric Nephrology
MassGeneral Hospital for Children
Boston, Massachusetts
Medical Genetics
MassGeneral Hospital for Children
Boston, Massachusetts
University of Wisconsin School of Medicine and Public Health
Madison, Wisconsin
Publication History
Initial Posting: September 12, 2013; Last Revision: December 15, 2022.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Tan W, Lin A, Keppler-Noreuil K. Cranioectodermal Dysplasia. 2013 Sep 12 [Updated 2022 Dec 15]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.

