NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
The NCBI Handbook [Internet]. 2nd edition. Bethesda (MD): National Center for Biotechnology Information (US); 2013-.
This publication is provided for historical reference only and the information may be out of date.
About Viral and Phage Genome Processing and Tools
Authors
Yiming Bao, PhD, 1 J. Rodney Brister, PhD,1 Olga Blinkova, PhD,1 Danso Ako-adjei, PhD,1 and Chetvernin Vyacheslav, PhD1.
1 J. Rodney Brister, PhD,1 Olga Blinkova, PhD,1 Danso Ako-adjei, PhD,1 and Chetvernin Vyacheslav, PhD1.Affiliations
Corresponding author.Created: March 30, 2013; Last Update: May 10, 2013.
Estimated reading time: 20 minutes
Scope
The National Center for Biotechnology Information (NCBI) Viral Genome Resource hosts all virus-related data and tools. All complete viral genome sequences deposited in the International Nucleotide Sequence Database Collaboration (INSDC) databases are collected by the NCBI Viral Genome Project (1). A RefSeq record is created from one of the complete genome sequences for each virus species, and the others are tagged as neighbors to the RefSeq. RefSeq records are subjected to curation procedures, which include automated gene locus_tag assignment, validation of molecule information and protein names, and annotation of novel proteins. Proteins encoded by RefSeqs are used to generate Protein Clusters, and the curated Protein Clusters are applied in turn to improve the annotation of new and existing RefSeqs. Sequence analyses such as global alignment of genome neighbors to RefSeq are provided. Databases specific to viruses that have a large number of genome sequences generated from sequencing projects are created for easy data retrieval and analyses. Tools that facilitate viral genome annotation and classification are also available.
History
Like other organisms, the number of sequences for viruses increased dramatically in recent years thanks to the advances of sequencing technologies. There are over 500,000 sequences in the INSDC databases for human immunodeficiency virus 1 alone. This makes it very difficult for researchers to efficiently work with these sequences directly from the databases. Also, many sequences are very short, which users may not want to include in their searches or analyses. So a collection of complete viral genome sequences is desired. Viruses are unique compared to other organisms in that there is significant variability in the forms of their genomes—linear or circular, single-stranded or double-stranded, DNA or RNA. The genome organization and expression strategy can vary dramatically from virus to virus. The taxonomic classification standards for some viruses are not very well established. All these factors contribute to sequence submission errors, such as wrong molecular information, incorrect/missing gene/protein annotation, and chaos of taxonomic assignment in some viral genome sequence records. As part of the NCBI’s Reference Sequence (RefSeq) database, the NCBI Viral Genome Project was created to cope with the issues described above.
Data Model
Viral Genome Reference Sequence and Genome Neighbors
From all complete viral genome sequences (including Viroids), one RefSeq record (or a set of RefSeq records for segmented viruses) is created for each species. Occasionally, more than one RefSeq record is created in a species to represent different subgroups of the virus (e.g., Dengue virus 1, 2, 3, and 4). All other complete viral genome sequences in the same species as the RefSeq become “neighbors” to the RefSeq. Both the RefSeq and neighbors can be retrieved from the NCBI’s Entrez database. Please note that genome neighbors are not the same as GenBank related sequences, which represent records selected by sequence similarity.
Virus Taxonomy
The Viral Genomes Project is tightly linked with the Taxonomy database. The names and classifications of viruses in the Taxonomy database follow, to a large extent, the most recent report of the International Committee on the Taxonomy of Viruses (ICTV, http://www.ictvonline.org). As the ICTV reports appear infrequently, the NCBI Taxonomy database attempts to stay current by also accepting new names and classification schemes on a case-by-case basis as provided in the reports of the ICTV executive meetings, taxonomic proposals approved by ICTV, and based on the advice of outside experts.
However, many sequence submissions are for viruses that are not listed in the ICTV report and sometimes not even described in the published literature. In spite of this, the taxonomy database can index these organisms and associated records, and these names are placed under an “unclassified” node to distinguish them from the ICTV-approved names.
Resources for Specific Viruses
Databases for specific viruses (e.g., Influenza, Dengue, and West Nile) that have a large number of genome sequences generated from sequencing projects are created for easy data retrieval and analyses. See the Virus Variation chapter for more information.
Databases such as the Coronavirus Resource are also made for viruses of medical importance.
Dataflow
Scan INSDC Databases for Complete Viral Genome Candidates
For the NCBI viral genome collection, a complete genome is one that contains all coding regions of the virus. Newly released viral sequences in the INSDC databases are constantly screened for complete genomes by an automated procedure. A sequence is considered a candidate for a complete genome if either of the following two criteria is met: (i) The topology of the sequence is circular or (ii) the definition of the sequence contains any of the following phrases: “complete genome”, “complete chromosome”, “sequence of the genome of”, or “complete genomic sequence”.
Some complete viral genome sequences are not detected by this automatic procedure because the source record either does not correctly indicate a circular topology or does not include the keywords listed above. To overcome this problem, viral sequences undergo an additional screening based on the sequence length. Only sequences longer than 85% of the length of the reference sequence in the species are selected as the complete genome candidates.
Additionally, complete viral genome sequences are identified with the aid of external scientific advisors, experts on particular families or groups of viruses, who also assist in the curatorial process. The list of advisors and their contact information is available at http://www.ncbi.nlm.nih.gov/genomes/GenomesHome.cgi?taxid=10239&hopt=advisors.
Accept Complete Viral Genome Candidates
NCBI staff manually review the complete viral genome candidates, and if satisfied, accept them as the reference sequence if there was not one in the species, or otherwise as neighbors to existing reference sequences.
When more than one complete genome is available for reference sequence, preference is given to the sequence of a well-studied and practically important virus isolate, and/or the one that has the best annotation.
The taxonomic classifications of the viruses where the new genomes are obtained are rigorously checked at this step. It is not uncommon that the GenBank submitter gives a sequence a new species name when it really belongs to an existing species. If the sequence is accepted without verification, an undesired reference sequence record will be created when it should actually be considered as a neighbor to another reference sequence. Finding an appropriate taxonomic position for a virus usually involves comparative sequence analysis, using tools like BLAST and PASC (described below). The ICTV Study Groups are frequently consulted for taxonomy issues. When in doubt, a complete genome candidate will have a “wait” status until the issues are resolved, in which case, there is a delay in the creation of the reference sequence.
Segmented Viruses
Segmented viruses are those with more than one genome component (segment). Candidates for complete sequences of individual segments are determined using similar criteria as the ones described above for single-component viruses. One sequence is selected for each segment to form a set of reference sequences that covers all segments of the genome. The reference genome set is manually assembled by matching strain and isolate information for available sequences of complete components. When several sequences are available for the same segment of the same strain and/or isolate, preference is given to a sequence obtained in the same laboratory as those of the other components. Other complete sequences become neighbors to the reference sequence of the same segment, and a segment name provided by NCBI staff is used to connect neighbors to the corresponding reference sequence.
RefSeq Creation
A RefSeq record is created from the INSDC sequence accepted by NCBI staff. Accession numbers unique to RefSeq records are assigned to the nucleotide (NC_XXXXXX) and protein (NP_XXXXXX, YP_XXXXXX or YP_XXXXXXXXX) sequences. Gene locus_tags are assigned to the RefSeq as well.
RefSeq Curation
The curatorial process includes the correction and update of the record, along with the addition of relevant biological information taken from the literature, other sequence records, original submitters, and outside advisors. The most common corrections are made to the type and topology of the genomic molecule (double strand or single strand, linear or circular) as well as to taxonomy lineage.
A large part of the curatorial process involves improvement of genome annotation, which includes searches for missing genes, assignment of functional roles to protein products, correction of annotations for proteins expressed by frame shifting or read through, restoration of proteins disrupted by sequencing errors, and addition of post translational processing information. Some RNA viruses encode polyproteins that contain multiple functional domains and are cleaved by proteinases into mature peptides. NCBI staff adds mature peptide annotation (from polyproteins) to viral RefSeq records if they were not present in the original INSDC records (compare NC_002532 and X53459). These mature peptides have RefSeq protein accession numbers and are thus indexed and retrievable as individual proteins.
RefSeq has established a number of collaborations in an effort to improve the accuracy of viral sequence records. In collaboration with Mark Borodovsky, the GeneMark program (http://exon.gatech.edu/VIOLIN) was used to predict open reading frames (ORFs) in some viral RefSeq genomes and to compare them with the original annotations. For example, the original GenBank record for the complete genomic sequence of a large double-stranded DNA virus—Sheeppox virus (AY077832) contained no protein annotations. Subsequently, 147 protein coding genes were predicted by the GeneMark program in the genome, and added to the corresponding RefSeq record (NC_004002).
Another ongoing collaboration project is the revision and annotation of overlapping genes. Gene overlaps, which can be defined as having nucleotides coding for more than one protein by being read in multiple reading frames, are a common feature of viruses (2). Proteins created by gene overlaps are typically accessory proteins that play a role in viral pathogenicity or spread (3, 4). Despite their importance, overlapping genes are difficult to identify and are often overlooked. Carefully annotated and curated data on overlapping genes in viral genome RefSeqs allow researchers to conduct studies on evolution and informational characteristics of overlapping genes as well as on functionality of corresponding products. With the help of Andrew Firth from the University of Cambridge, Cambridge, UK, and David Karlin from the University of Oxford, Oxford, UK, we have been working on adding (or correcting) missing overlapping genes and corresponding proteins in virus RefSeqs. At the present time, at least one RefSeq representative for each genus from the 14 selected virus families (Arteriviridae, Arteriviridae, Bunyaviridae, Caliciviridae, Circoviridae, Disistroviridae, Flavoviridae, Luteoviridae, Paramixovridae, Parvoviridae, Picornaviridae, Potyviridae, Reoviridae, Togaviridae) was corrected based on experimental or predictive analysis. For each new protein the position of start and end codon is determined based on the experimental data or according to comparative analysis described in the literature. Protein names, their functions (if known), experimental data and literature links are added for each protein. The frameshifting sites (if present) and the nature of a frameshift are added to the genome annotations based on the most recent literature data. RefSeq NC_001479 is an example of a sequence with recently discovered gene overlaps. It represents the encephalomyocarditis virus (EMCV) species from the family Picornaviridae. According to the experimental analysis performed by Loughran et al. (5) a conserved ORF overlaps the 2B-encoding sequence of EMCV in the +2 reading frame. A previously overlooked ORF is translated as a 128-129 amino acid transframe fusion (2B*) with the N-terminal 11-12 amino acids of 2B, via ribosomal frameshifting. To represent the results of this study, we added the truncated version of polyprotein (CDS positions: 834-3998, 3998-4351) and 2B* protein (mature peptide with the coding region positions: 3966-3998, 3998-4348). Another example of overlapping genes is RefSeq NC_008311 belonging to the Murine norovirus (MNV) species from the family Caliciviridae. We updated the annotation to add a recently discovered (6) virulence factor 1 protein (VF1) encoded by subgenomic RNA (CDS coordinates: 5069-5710) in an alternative reading frame overlapping the VP1 coding region.
RefSeq proteins are clustered on the basis of sequence homology within the Protein Clusters resource and curated in aggregate by NCBI staff (also see the Protein Clusters chapter). This curation includes the assignment of functional protein names to clusters that can in turn be propagated to individual protein records in RefSeq, yielding consistent, informative names among clustered proteins. RefSeq staff work in collaboration with a number of stakeholders including SwissProt, ICTV, sequencing centers, and scientific communities to develop annotation and protein naming standards. The goal is to improve the quality and consistency of viral genome annotation in both RefSeq and INSDC databases.
HIV-1, Human Protein Interaction Database
Although numerous advances have been made in the fields of retrovirology and AIDS research, much of the biological processes that underlie infection, replication, and immune evasion are unknown. Similarly, the mechanisms that orchestrate cellular restriction to infection as well as those that potentiate the innate and adaptive immune systems are poorly understood. The human immunodeficiency virus type 1 (HIV-1) RNA genome (NC_001802) encodes three major genes from which the major proteins—group specific antigen (Gag), polymerase (Pol), and envelope (Env)—are transcribed. Through various combinations of overlapping reading frames, differential splicing, and proteolytic cleavage, several HIV-1 proteins with regulatory and auxiliary roles are also expressed. These include the proteins transactivator (Tat), regulator of viral protein expression (Rev), negative factor (Nef), viral protein R (Vpr), viral infectivity factor (Vif), and viral protein U (Vpu). Tat and Rev regulate transcription and HIV-1 nuclear RNA export, respectively, while the accessory proteins Nef, Vpr, Vif, and Vpu are dispensable for replication in certain cell types (7).
A large number of protein-protein interactions involving viral and cellular proteins are required for both cellular immunity and competent viral infection. Information about protein-protein interactions is critical to advancements in vaccine research, therapeutic drug discovery, and cell biology. In order to facilitate these advancements, the HIV-1, Human Protein Interaction Database, was established to catalog all data published in peer-reviewed journals regarding HIV-1 and human protein interactions. Included in this database are brief descriptions of the respective interactions, National Library of Medicine (NLM) PubMed identification numbers (PMIDs) for articles describing the interaction, NCBI Reference Sequence (RefSeq) protein accession numbers, NCBI Entrez Gene ID numbers, and keywords that facilitate interactions searches.
The database is organized in a fashion that provides downloadable or onsite-viewable reports for the HIV-1 proteins. The protein interactions are categorized by 43 interaction keywords including binds, cleaved by, degrades, stimulates, co-localizes with, and recruits. By utilizing these keywords in drop-down menus, a user can narrow down a search for particular interaction types for specific HIV-1 proteins. For instance, the viral protein, Vif, binds the mammalian cellular protein apolipoprotein B mRNA editing enzyme (APOBEC3G) and targets it for proteasomal destruction. In the absence of Vif, APOBEC3G incorporation into HIV-1 virions leads to G-to-A hypermutation of the viral genome and markedly reduced replicative potential (8). By clicking on “vif” and then using the drop-box to choose “degrades” and hitting the “view” button, a report containing APOBEC3G and several similar interactions can be obtained. These reports can then be downloaded as a text file if the user chooses. Because the HIV-1 interaction data is integrated into the Entrez Gene database, information about protein domain structure, genomic context, synonymous names, gene loci, and links to order clones of the human genes may be also obtained. The availability of resources such as this can provide insights into the many biological processes that involve HIV-1 infection, replication, and evolution. Likewise, they provide data that may one day permit predictive modeling and/or construction of structural interaction networks (9).
This database is made available through the National Library of Medicine at http://www.ncbi.nlm.nih.gov/RefSeq/HIVInteractions (10) and a set of all the current interactions can be obtained by FTP from ftp://ftp.ncbi.nih.gov/gene/GeneRIF under the file, hiv_interactions.gz.
Access
Viral Genome Resource Homepage
All NCBI virus-related data can be accessed through the Viral Genome Resource homepage (http://www.ncbi.nlm.nih.gov/genomes/GenomesHome.cgi?taxid=10239). On the left hand side bar, users can find links to information about the viral genome resource, list of viral genomes in alphabetical order or by taxonomic groups, viral RefSeq genome and protein records, sequence records of genome neighbors, ftp site for viral RefSeq data, virus-related tools, and resources for specific viruses. The main page lists large groups of viruses, which can be opened and moved down to lower level taxonomic lineages that contain RefSeqs. A search box is also provided for quick location of viral genomes belonging to a particular group (e.g., family).
The Viral Genome Presentation
The top level Viral Genome Presentation lists all viruses that have RefSeqs and is available at http://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=10239. A list of the next level sub-lineages (e.g., ssRNA viruses) is provided so users can easily move down the taxonomic hierarchy to find viral genomes of their interest. An example viral genome presentation page for Potyviridae (taxid=39729) is shown in Figure 1.
Global Alignment of Genome Neighbors
Pairwise global alignment is performed between each genome neighbor and their corresponding RefSeq, using the "band" version of the Needleman-Wunsch algorithm. A graphical representation of the alignment is available when the number under the “Nbrs” column on viral genome presentation pages (Figure 1C) is clicked. For segmented viruses, the alignments for each genome segment are sequentially displayed. An example of the graphical view is shown in Figure 2.
The Genome Browser
Viral RefSeqs are also presented with all genome records of other organisms in the NCBI Genome Browser (http://www.ncbi.nlm.nih.gov/genome/browse), which can be filtered by large groups (dsDNA viruses, ssRNA viruses, etc.), subgroups (mostly viral families), and hosts.
Viral Genome Data Through ftp
As part of the bi-monthly RefSeq releases, all nucleotide and protein sequences and GenBank flat files for viral genomes are available at ftp://ftp.ncbi.nih.gov/refseq/release/viral. Various format of genome data for individual viruses are available at ftp://ftp.ncbi.nlm.nih.gov/genomes/Viruses/.
Related Tools
FLAN
The influenza virus genome annotation tool (FLAN, for FLu ANnotation) was created as a result of NCBI’s participation in the Influenza Genome Sequencing Project (11) which was initiated by the National Institute of Allergy and Infectious Diseases. Under this project, influenza virus samples provided by collaborators worldwide are sequenced by the J Craig Venter Institute, submitted to NCBI for genome annotation, and released immediately in GenBank. Since the beginning of this project in 2005, more than 11,000 genomes of influenza viruses have been sequenced and published in GenBank. Because of the large number of sequences, an automated genome annotation pipeline is required.
FLAN is an application for user-provided Influenza A virus, Influenza B virus, and Influenza C virus sequences. It can predict protein sequences encoded by a flu sequence and produce a feature table that can be used for sequence submission to GenBank, as well as a GenBank flat file.
The type/segment/subtype of an input influenza sequence is first determined by BLAST, and then aligned against a corresponding reference protein set with a "Protein to nucleotide alignment tool"—ProSplign (http://www.ncbi.nlm.nih.gov/sutils/static/prosplign/prosplign.html). The translated product from the best alignment to the sample protein sequence is used as the predicted protein encoded by the input sequence.
In addition to the creation of the feature table, FLAN can also determine and report the following properties of flu sequences: influenza virus species (A, B, or C), length, genome segment, subtype of the HA and NA segment, common drug-resistant mutations in segment NA and M, mutations in the PB2 segment that might confer high virulence of influenza viruses, possible contaminated/vector sequences at the ends, the completeness of the nucleotide, protein and coding regions, insertion/deletion that will disrupt the coding regions, and premature stop codons in the coding regions. These capabilities of FLAN are used to populate certain fields in the Influenza Virus Sequence Database (12). They also make FLAN a useful tool for flu sequence validation to identify possible sequencing errors or human errors in segment/subtype assignment.
Internally, FLAN is implemented in a NCBI-developed framework which allows the execution of background CGI tasks for more than 30 s (default WEB frontend timeout). This allows the online interface of FLAN to process hundreds of flu sequences at a time.
In an effort to maintain consistent and high quality annotations of flu sequences, FLAN is recommended by GenBank as the tool to generate feature tables that can be used for flu sequence submissions to GenBank through the recently implemented “virus wizard” in Sequin.
FLAN (13) is available at http://www.ncbi.nlm.nih.gov/genomes/FLU/Database/annotation.cgi.
PASC
Viruses are classified based on their properties such as morphology, serology, host range, genome organization, and sequence. The dramatic increase of virus sequences in public databases makes sequence-based virus classification more feasible.
The most commonly used virus classification tool based on sequence is phylogenetic analysis. The classification of about 70% of families and floating genera described in the ninth Report of ICTV are supported by phylogenetic trees (14). Despite its wide-spread use, phylogenetic analysis is usually computationally intensive, and requires expertise to interpret the results.
Recently, a novel method using natural vector based on the distributions of nucleotide sequences was reported on virus classifications (15).
Another sequence-based molecular classification method for viruses is to calculate pairwise identities of virus sequences within a virus family, and the number of virus pairs at each percentage is plotted. This will usually produce peaks that represent different taxonomic groups such as variants, species, and genera, and the percentages at borderlines of the peaks can be used as demarcation criteria for different taxa. This method has been applied to a few viral families including Coronaviridae (16), Geminiviridae (17), Papillomoviridae (18), Picornaviridae (19), and Potyviridae (20). A major drawback of this method is the inconsistency of the results when different protocols are used to calculate the pairwise identities. It is very difficult, if not impossible, for researchers to use the exact algorithm and parameters to test their own sequences as those used to establish the demarcation criteria (usually) by others. The identities obtained from the two systems are therefore not comparable. To overcome this problem, NCBI created the PASC (Pairwise Sequence Comparison) resource (21), where the same protocol is used for both the establishment of the demarcation criteria and the testing of new viral sequences. Many viral groups were included in the resource.
For a given virus family/group, complete genome sequences are retrieved from the NCBI viral genomes collection described in this chapter, which include both RefSeqs and neighbors. These sequences, together with their lineages in the NCBI Taxonomy database, are stored in a database. The database is updated every day to add new genome sequences and reflect taxonomy changes.
Traditionally, genome identities were calculated based on pairwise global alignments in PASC. Although this method works well for some virus families/groups such as papillomaviruses and potyviruses, the results are not optimized for others mainly for the following reasons:
- 1.
In some viruses with circular genomes such as the circoviruses, there is an inconsistency in the designation of the first nucleotide of the genome sequences in public databases.
- 2.
In some viruses, particularly those with negative-strand RNA genomes, the opposite strand of the of the genome are sometimes submitted to the public databases. When genome identities are calculated based on the global alignment of two genomes in the opposite strand, the result is usually lower than what they should be.
- 3.
For viruses that are distantly related, the identities obtained by global alignment are usually misleading, because the minimum identity of any two random genome sequences of the same size is 25%.
To overcome these issues, a BLAST-based alignment method is used. Two sets of BLAST (22) are performed on each pair of genome sequences. In the first set, the translated protein sequences of one genome in six frames are searched against the nucleotide sequence of the other genome using tblastn. The amino acid alignments in the tblastn results are converted back to nucleotide alignments. In the second BLAST set, pairwise blastn is carried out on the nucleotide sequences of the genomes. We then select a consistent set of hits from the two sets of BLAST results, preferring higher identity hits and trimming overlaps out of lower identity hits. This process will select blastn hits for close genomes, but most likely tblastn hits for distant ones. A mixture of blastn and tblastn hits might be used in some cases. Pairwise identities are calculated as the total number of identical bases in local hits divided by the mean sequence length of the genome pair. This method greatly improves the performance of PASC in some virus families (see Figure 3 for an example).
The identity distribution chart is plotted based on pairwise alignments computed between every member of the selected virus family or group. The pair is represented in green color if both genomes belong to the same species according to their assignment in NCBI’s Taxonomy database; in yellow color if the two genomes belong to different species but the same genus; and in peach color if they belong to different genera. Both linear and log scales are available for the Y-axis (number of pairs).
To compare external genomes against existing ones in the database, specify the query genomes in the "Sequence" box, using either their GenBank Accession/GI numbers, entering the raw sequence in FASTA format, or uploading a file containing the sequences by clicking the "Browse" button. Up to 25 sequences can be added in one submission. After sequences are submitted, PASC will start computing pairwise identities between user-provided genomes and the existing genome sequences of the family. At the end of the process, for each input genome, PASC produces a list of pairwise identities, from the highest to the lowest, between this input genome and 1) the rest of input genomes (if there are more than one), and 2) 5 to 10 closest matches to existing genomes within the family. The identity distribution chart will depict the currently selected genome with a different color. One can click on each genome's number to make it current, or can click the identity to see details of the alignment.
PASC can be used to:
- 1.
Establish demarcation criteria for taxonomic classification of certain viruses, as demonstrated in the Filoviridae family (23).
- 2.
Identify viruses that were incorrectly assigned in the taxonomy database.
- 3.
Classify viruses with newly sequenced genomes.
PASC can be accessed through http://www.ncbi.nlm.nih.gov/sutils/pasc. It currently covers more than 52 virus families/groups, which are listed at http://www.ncbi.nlm.nih.gov/sutils/pasc/viridty.cgi?textpage=main.
Genotyping Tool
The retroviral family, Retroviridae, is composed of numerous enveloped RNA viruses from which scientific study has revealed many interesting biological principles. Because of both its historical and present health implications, the human immunodeficiency virus 1 (HIV-1) has been of major interest in the scientific and medical communities. As a result, the ability to quickly and efficiently identify HIV genotypes is critical to several areas of scientific and medical research. For instance, because of the almost unavoidable trend of HIV-1 drug resistance in infected individuals, the medical treatment of HIV-1 infected individuals is particularly driven by genotypic studies (24). Likewise drug discovery trials and epidemiological studies are also motivated by similar concerns.
By comparisons to preexisting alignments and trees, phylogenetic analysis can be used to discriminate between viral genotypes as well as to determine the subtype of new isolates. This can pose a particular problem with respect to viruses as coinfection and superinfection leading to inter-subtype recombination is not entirely uncommon (25). Because phylogenetic analyses cannot always distinguish such recombinants and new subtypes, several methods that analyze segments of the genome have been designed (26). The high selective pressure, and high error and replication rate of RNA viruses such as HIV-1 often makes it difficult to align viral sequences automatically.
The NCBI Genotyping tool (27), utilizes an algorithm that uses scored BLAST (22) pairwise alignments between overlapping segments of the query and reference sequences for each virus. The algorithm uses a “sliding window” along a query sequence that processes each window-sequence and segment separately. By comparing each segment to a set of reference sequences from BLAST-derived analyses, a similarity scores for each local alignment is obtained. Each query segment is assigned the reference sequence genotype that matches the query with the highest BLAST similarity score. This process is repeated for every subsequent “window” in the same manner until the entirety of the query sequence is covered by overlapping BLAST alignments. The results from all windows are combined and displayed graphically. By displaying the results of multiple segments in a computer generated graphical format, it is easier for the end-user to determine the genotype of a query sequence. Likewise, because of the manner in which the results are obtained, recombinant genotypes and recombination breakpoints can also be identified. Currently, the NCBI Genotyping tool utilizes reference sets from HIV-1, hepatitis B virus (HBV), hepatitis C virus (HCV), human T-lymphotropic virus 1 and 2 (HTLV-1 and HTLV-2), simian immunodeficiency virus (SIV) and poliovirus (PV). The tool is located at http://www.ncbi.nlm.nih.gov/projects/genotyping/formpage.cgi.
References
- 1.
- Bao Y, Federhen S, Leipe D, Pham V, Resenchuk S, Rozanov M, Tatusov R, Tatusova T. National center for biotechnology information viral genomes project. J Virol. 2004 Jul;78(14):7291–8. [PMC free article: PMC434121] [PubMed: 15220402]
- 2.
- Rancurel C, Khosravi M, Dunker AK, Romero PR, Karlin D. Overlapping genes produce proteins with unusual sequence properties and offer insight into de novo protein creation. J Virol. 2009 Oct;83(20):10719–36. [PMC free article: PMC2753099] [PubMed: 19640978]
- 3.
- Chirico N, Vianelli A, Belshaw R. Why genes overlap in viruses. Proc Biol Sci. 2010 Dec 22;277(1701):3809-17. [PMC free article: PMC2992710] [PubMed: 20610432]
- 4.
- Sabath N, Wagner A, Karlin D. Evolution of viral proteins originated de novo by overprinting. Mol Biol Evol. 2012 Dec;29(12):3767–80. [PMC free article: PMC3494269] [PubMed: 22821011]
- 5.
- Loughran G, Firth AE, Atkins JF. Ribosomal frameshifting into an overlapping gene in the 2B-encoding region of the cardiovirus genome. Proc Natl Acad Sci U S A. 2011 Nov 15;108(46):E1111–9. [PMC free article: PMC3219106] [PubMed: 22025686]
- 6.
- McFadden N, Bailey D, Carrara G, Benson A, Chaudhry Y, Shortland A, Heeney J, Yarovinsky F, Simmonds P, Macdonald A, Goodfellow I. Norovirus regulation of the innate immune response and apoptosis occurs via the product of the alternative open reading frame 4. PLoS Pathog. 2011 Dec;7(12):e1002413. [PMC free article: PMC3234229] [PubMed: 22174679]
- 7.
- Hughes, SH, Varmus, HE. Retroviruses. New York: CSHL Press; 1997. [PubMed: 21433340]
- 8.
- Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003 Nov;9(11):1404–7. [PubMed: 14528300]
- 9.
- Franzosa EA, Garamszegi S, Xia Y. Toward a three-dimensional view of protein networks between species. Front Microbiol. 2012;3:428. [PMC free article: PMC3528071] [PubMed: 23267356]
- 10.
- Fu W, Sanders-Beer BE, Katz KS, Maglott DR, Pruitt KD, Ptak RG. Human immunodeficiency virus type 1, human protein interaction database at NCBI. Nucleic Acids Res. 2009 Jan;37(Database issue):D417–22. [PMC free article: PMC2686594] [PubMed: 18927109]
- 11.
- Ghedin E, Sengamalay NA, Shumway M, Zaborsky J, Feldblyum T, Subbu V, Spiro DJ, Sitz J, Koo H, Bolotov P, Dernovoy D, Tatusova T, Bao Y, St George K, Taylor J, Lipman DJ, Fraser CM, Taubenberger JK, Salzberg SL. Large-scale sequencing of human influenza reveals the dynamic nature of viral genome evolution. Nature. 2005 Oct 20;437(7062):1162–6. [PubMed: 16208317]
- 12.
- Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. The influenza virus resource at the National Center for Biotechnology Information. J Virol. 2008 Jan;82(2):596–601. [PMC free article: PMC2224563] [PubMed: 17942553]
- 13.
- Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Tatusova T. FLAN: a web server for influenza virus genome annotation. Nucleic Acids Res. 2007 Jul;35(Web Server issue):W280-4. [PMC free article: PMC1933127] [PubMed: 17545199]
- 14.
- King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. Virus taxonomy—ninth report of the International Committee on Taxonomy of viruses. London: Elsevier/Academic Press; 2011.
- 15.
- Yu C, Hernandez T, Zheng H, Yau SC, Huang HH, He RL, Yang J, Yau SS. Real time classification of viruses in 12 dimensions. PLoS One. 2013 May 22;8(5):e64328. [PMC free article: PMC3661469] [PubMed: 23717598]
- 16.
- González JM, Gomez-Puertas P, Cavanagh D, Gorbalenya AE, Enjuanes L. A comparative sequence analysis to revise the current taxonomy of the family Coronaviridae. Arch Virol. 2003 Nov;148(11):2207–35. [PMC free article: PMC7087110] [PubMed: 14579179]
- 17.
- Fauquet CM, Briddon RW, Brown JK, Moriones E, Stanley J, Zerbini M, Zhou X. Geminivirus strain demarcation and nomenclature. Arch Virol. 2008;153(4):783–821. [PubMed: 18256781]
- 18.
- Bernard HU, Burk RD, Chen Z, van Doorslaer K, Hausen H, de Villiers EM. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology. 2010 May 25;401(1):70–9. [PMC free article: PMC3400342] [PubMed: 20206957]
- 19.
- Oberste MS, Maher K, Kilpatrick DR, Pallansch MA. Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J Virol. 1999 Mar;73(3):1941–8. [PMC free article: PMC104435] [PubMed: 9971773]
- 20.
- Adams MJ, Antoniw JF, Fauquet CM. Molecular criteria for genus and species discrimination within the family Potyviridae. Arch Virol. 2005 Mar;150(3):459–79. [PubMed: 15592889]
- 21.
- Bao Y, Kapustin Y, Tatusova T. Virus Classification by Pairwise Sequence Comparison (PASC). In: Mahy BWJ, Van Regenmortel MHV, Editors. Encyclopedia of Virology, 5 vols. Oxford: Elsevier; 2008. Vol. 5, 342-348.
- 22.
- Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997 Sep 1;25(17):3389–402. [PMC free article: PMC146917] [PubMed: 9254694]
- 23.
- Bao Y, Chetvernin V, Tatusova T. PAirwise Sequence Comparison (PASC) and Its Application in the Classification of Filoviruses. Viruses. 2012 Aug;4(8):1318–27. [PMC free article: PMC3446765] [PubMed: 23012628]
- 24.
- Gallant, JE. "Antiretroviral drug resistance and resistance testing." Topics in HIV medicine: a publication of the International AIDS Society, USA. 2005;13.5:138. [PubMed: 16377851]
- 25.
- Ramos A, Hu DJ, Nguyen L, Phan KO, Vanichseni S, Promadej N, Choopanya K, Callahan M, Young NL, McNicholl J, Mastro TD, Folks TM, Subbarao S. Intersubtype human immunodeficiency virus type 1 superinfection following seroconversion to primary infection in two injection drug users. J Virol. 2002 Aug;76(15):7444–52. [PMC free article: PMC136380] [PubMed: 12097556]
- 26.
- Siepel AC, Halpern AL, Macken C, Korber BT. A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences. AIDS Res Hum Retroviruses. 1995 Nov;11(11):1413–6. [PubMed: 8573400]
- 27.
- Rozanov M, Plikat U, Chappey C, Kochergin A, Tatusova T. A web-based genotyping resource for viral sequences. Nucleic Acids Res. 2004 Jul 1;32 (Web Server issue):W654-9. [PMC free article: PMC441557] [PubMed: 15215470]
Figures
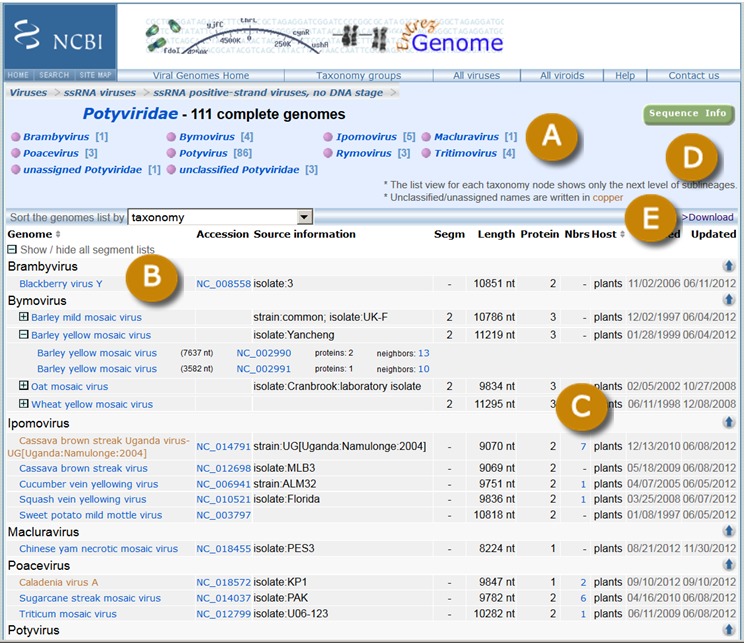
Figure 1.
Viral genome collections in the family Potyviridae. A. A list of sub-lineages one level below the Potyviridae node in NCBI’s Taxonomy database. The numbers in square brackets represent the number of genomes within the taxonomic node. B. Organism names of viral genomes. The ICTV approved names are in blue, and the unassigned/unclassified ones are in cooper. C. The number of genome neighbors to the RefSeq. The numbers are linked to the global alignment of genome neighbors with the RefSeq (Figure 2). D. Links to show all RefSeq nucleotide or protein records for the viruses displayed. E. Links to download a table with the information displayed or a list of accession numbers of the RefSeq records.
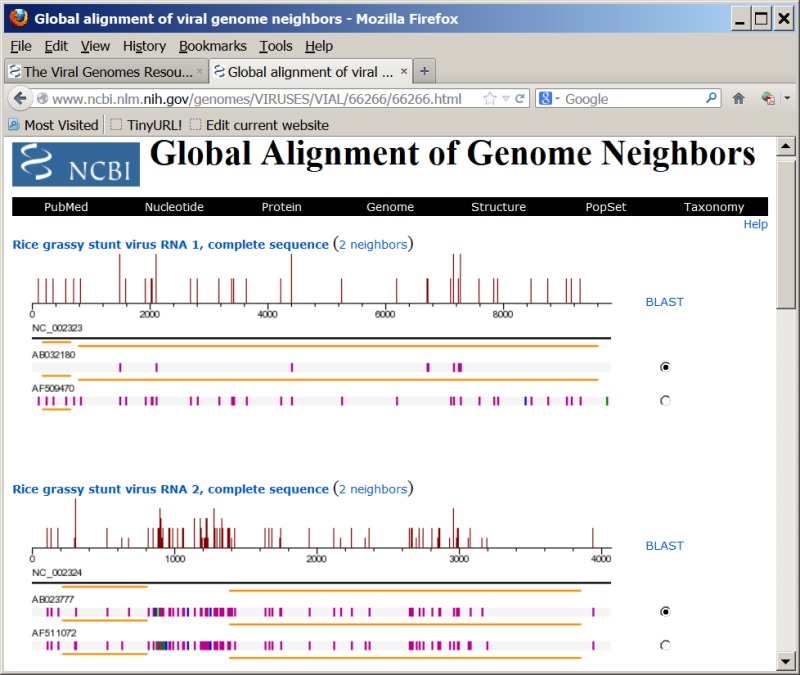
Figure 2.
Global alignment of genome neighbors with the RefSeq of Rice grassy stunt virus (NC_002323 and NC_002324). The magenta, blue, and green bars represent differences, deletions, or insertions in sequences, compared with the reference sequence. The orange lines represent coding regions annotated in the genome records. The histogram shows the average density of nucleotide changes (excluding gaps, insertions, and undetermined nucleotides) in all genome neighbors for each reference sequence segment.
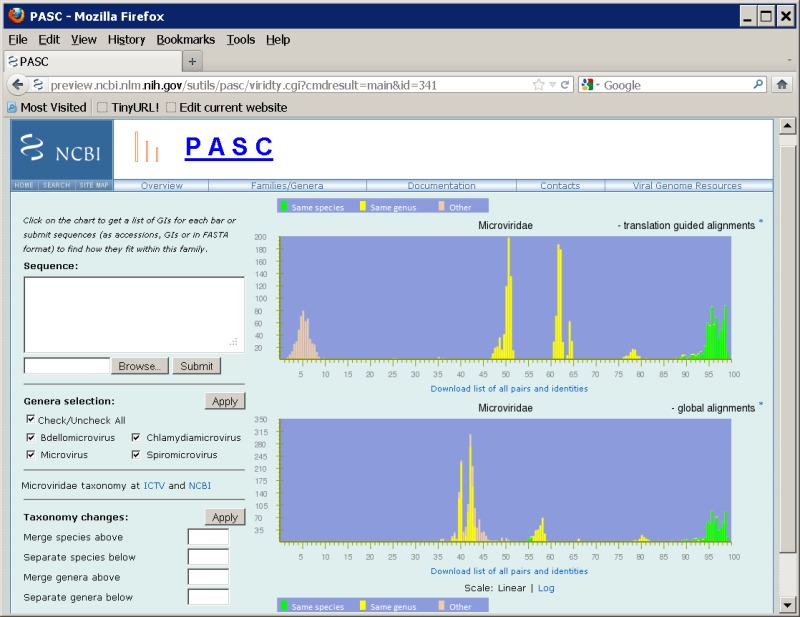
Figure 3.
Frequency distribution of pairwise identities from the complete genome sequence comparison of 71 microviruses.