Summary
Clinical characteristics.
COMP-related pseudoachondroplasia (COMP-PSACH) is characterized by normal length at birth and normal facies. Often the presenting feature is a waddling gait, recognized at the onset of walking. Typically, the growth rate falls below the standard growth curve by approximately age two years, leading to a moderately severe form of disproportionate short-limb short stature. Joint pain during childhood, particularly in the large joints of the lower extremities, is common. Degenerative joint disease is progressive; approximately 50% of individuals with COMP-PSACH eventually require hip replacement surgery.
Diagnosis/testing.
The diagnosis of COMP-PSACH can be made on the basis of clinical findings and radiographic features. Identification of a heterozygous pathogenic variant in COMP on molecular genetic testing establishes the diagnosis if clinical features are inconclusive.
Management.
Treatment of manifestations: Analgesics for joint pain; encourage physical activities that do not cause excessive wear and/or damage to the joints; osteotomy for lower limb malalignment; rarely, surgery for scoliosis; C1-C2 fixation for symptoms and radiographic evidence of cervical spine instability; attention to and social support for psychosocial issues related to short stature for affected individuals and their families.
Surveillance: Assess growth at each visit throughout childhood. Regular examinations for evidence of symptomatic joint hypermobility and/or lower limb malalignment, kyphoscoliosis, degenerative joint disease, and neurologic manifestations, particularly spinal cord compression secondary to odontoid hypoplasia. Assess for psychosocial issues annually or at each visit.
Agents/circumstances to avoid: In those with odontoid hypoplasia, extreme neck flexion and extension should be avoided.
Genetic counseling.
COMP-PSACH is inherited in an autosomal dominant manner. Some individuals diagnosed with COMP-PSACH have an affected parent. A proband diagnosed with COMP-PSACH may have the disorder as the result of a de novo pathogenic variant. Each child of an individual with COMP-PSACH and a reproductive partner with normal bone growth has a 50% chance of inheriting the COMP pathogenic variant and having COMP-PSACH. Because many individuals with short stature select reproductive partners with short stature, offspring of individuals with COMP-PSACH may be at risk of having double heterozygosity for two dominantly inherited bone growth disorders. Once the COMP pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
COMP-related pseudoachondroplasia (COMP-PSACH) should be suspected in individuals with the following clinical findings and radiographic features.
Clinical findings
- Normal length at birth
- Normal facies
- Waddling gait, recognized at the onset of walking
- Decline in growth rate to below the standard growth curve by approximately age two years, leading to moderately severe disproportionate short-limb short stature
- Moderate brachydactyly
- Ligamentous laxity and joint hyperextensibility, particularly in the hands, knees, and ankles
- Mild myopathy reported for some individuals
- Restricted extension at the elbows and hips
- Valgus, varus, or windswept deformity of the lower limbs
- Mild scoliosis
- Lumbar lordosis (~50% of affected individuals)
- Joint pain during childhood, particularly in the large joints of the lower extremities; may be the presenting symptom in mildly affected individuals
Radiographic features
- Delayed epiphyseal ossification with irregular epiphyses and metaphyses of the long bones (consistent)
- Small capital femoral epiphyses, short femoral necks, and irregular, flared metaphyseal borders; small pelvis and poorly modeled acetabulae with irregular margins that may be sclerotic, especially in older individuals
- Significant brachydactyly; short metacarpals and phalanges that show small or cone-shaped epiphyses and irregular metaphyses; small, irregular carpal bones
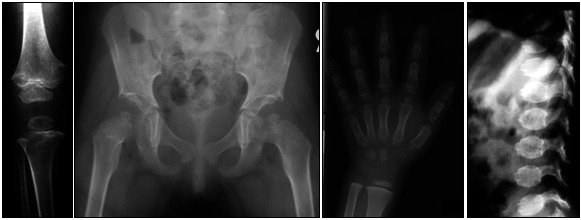
- Anterior beaking or tonguing of the vertebral bodies on lateral view. This distinctive appearance of the vertebrae normalizes with age, emphasizing the importance of obtaining in childhood the radiographs to be used in diagnosis (see Figure 1).
Figure 1.
Radiographs of a prepubertal child showing the changes typical of pseudoachondroplasia
Establishing the Diagnosis
The diagnosis of COMP-PSACH is established in a proband with the abovementioned clinical and radiographic features. The diagnosis is ideally confirmed on radiographs obtained in prepubertal individuals. At a minimum, AP views of the hips, knees, hands, and wrists and a lateral view of the spine are required (see Figure 1). Identification of a heterozygous pathogenic (or likely pathogenic) variant in COMP by molecular genetic testing (see Table 1) establishes the diagnosis if clinical features are inconclusive.
Note: Per ACMG/AMP variant interpretation guidelines, the terms "pathogenic variant" and "likely pathogenic variant" are synonymous in a clinical setting, meaning that both are considered diagnostic and can be used for clinical decision making [Richards et al 2015]. Reference to "pathogenic variants" in this GeneReview is understood to include likely pathogenic variants.
Molecular genetic testing approaches can include a single-gene testing or use of multigene panel:
- Single-gene testing. Sequence analysis of COMP is performed first to detect missense, nonsense, and splice site variants and small intragenic deletions/insertions. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
- A multigene panel that includes COMP and other genes of interest (see Differential Diagnosis) may be considered to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Table 1.
Molecular Genetic Testing Used in COMP-Related Pseudoachondroplasia
Clinical Characteristics
Clinical Description
COMP-related pseudoachondroplasia (COMP-PSACH) is characterized by disproportionate short-limb short stature. Intrafamilial and interfamilial variability are observed. Natural history is well documented [Wynne-Davies et al 1986, McKeand et al 1996]. To date, more than 500 individuals have been identified with COMP-PSACH. The following description of the phenotypic features associated with this condition is based on these reports.
Growth. Affected individuals are generally of normal length at birth. Typically, the growth rate falls below the standard growth curve by approximately age two years, leading to moderately severe disproportionate short-limb short stature. Growth curves for COMP-PSACH have been developed [Horton et al 1982]. Mean adult height is 116 cm for females and 120 cm for males [McKeand et al 1996].
Facies. Head size and shape are normal, without dysmorphic features.
Gait. Often the presenting feature is a waddling gait, recognized at the onset of walking.
Extremities. COMP-PSACH is a short-limb form of dwarfism with all limb segments equally affected (rhizo-, meso-, and acromelia). Individuals have moderate brachydactyly. Ligamentous laxity and joint hyperextensibility is present particularly in the hands, knees, and ankles. Extension at the elbows may be limited, and the elbows and knees may appear large. Valgus, varus, or windswept deformity of the lower limbs is common.
Some individuals have mild myopathy.
Scoliosis/lordosis can be observed in childhood and may persist into adulthood; severity is variable and there are no effective treatments.
Osteoarthritis of the upper extremities and the spine may occur in early adult life. Degenerative joint disease is progressive, and approximately 50% of individuals with COMP-PSACH eventually require hip replacement surgery.
Odontoid hypoplasia is not a common finding but does sometimes occur. Cervical spine instability can result, but C1-C2 fixation is not generally necessary.
Genotype-Phenotype Correlations
A systematic analysis of the relationship between genotype and phenotype has been performed on 300 reported COMP pathogenic variants resulting in PSACH and/or autosomal dominant multiple epiphyseal dysplasia (MED) [Briggs et al 2014]. The following are correlations from this study. (For repeat and domain structure, see Molecular Pathogenesis.)
- Pathogenic missense variants of nucleotides encoding either the N- or C-type motifs within each of the type III calcium-binding domains showed no significant association with either the MED or the PSACH phenotype.
- Pathogenic missense variants in nucleotides encoding the fourth and fifth (of 8 total) type III calcium-binding repeats (i.e., T34 and T35) showed significant association with the MED compared to the PSACH phenotype.
- Pathogenic missense variants in nucleotides encoding the sixth through eighth type III calcium-binding repeats (i.e., T36, T37, and T38) were significantly associated with the PSACH phenotype.
- The majority of pathogenic in-frame deletions, insertions, or indels were associated with PSACH (n=74; 82%), whereas a smaller proportion were associated with MED (n=16; 18%); however, in several instances, the same pathogenic variant was reported to cause both PSACH and MED [Briggs et al 2014].
Correlations from prior studies:
- Individuals with a pathogenic variant in the seventh type III calcium-binding repeat are reported to have more severe short stature than those with pathogenic variants in the other type III repeats [Mabuchi et al 2003].
- Individuals heterozygous for the common p.Asp473del pathogenic variant, present in approximately 30% of affected individuals, have a consistent, typical form of PSACH and are severely short [Mabuchi et al 2003]. In contrast, the insertion of an adjacent Asp (GAC) codon (p.Asp473dup) results in mild MED [Délot et al 1999, Zankl et al 2007, Jackson et al 2012].
- Most type III calcium-binding repeats have both an N- and C-type motif (see Molecular Genetics). Specific missense variants that result in PSACH (as opposed to MED) affect residues in the C-type motif, whereas missense variants in the N-type motif generally result in MED [Jackson et al 2012]. In-frame deletions are found equally between the N-type and C-type motifs [Jackson et al 2012] and can cause both PSACH and MED.
Penetrance
Penetrance is 100%.
Nomenclature
In the past, four subtypes of pseudoachondroplasia, including dominant and recessive forms, were recognized under the term pseudoachondroplasia. The current classification recognizes a single, dominantly inherited phenotype referred to as COMP-related pseudoachondroplasia [Unger et al 2023].
COMP-PSACH was referred to as pseudoachondroplastic dysplasia in older literature.
Prevalence
No firm data on the prevalence of COMP-PSACH are available; it is estimated at 1:30,000 (see MedlinePlus).
Genetically Related (Allelic) Disorders
Autosomal dominant multiple epiphyseal dysplasia (MED) (see Differential Diagnosis for clinical description). Approximately 50% of individuals with molecularly confirmed autosomal dominant MED are heterozygous for a COMP pathogenic variant. As in COMP-related pseudoachondroplasia, the pathogenic variants causing autosomal dominant MED have been found in the exons encoding the type III binding repeats (~85%) and the carboxyl-terminal globular domain (~15%).
Differential Diagnosis
Table 2.
Genes of Interest in the Differential Diagnosis of COMP-Related Pseudoachondroplasia
Other forms of spondyloepimetaphyseal dysplasia (SEMD). Many different skeletal dysplasias have abnormalities of the spine, metaphyses, and epiphyses apparent on radiographs. For example, Spranger et al [2005] described a severe form of SEMD with some radiographic similarity to COMP-related pseudoachondroplasia (COMP-PSACH) but without a COMP pathogenic variant. Generally, a complete genetic skeletal survey can distinguish these phenotypes from COMP-PSACH.
Another resource to help diagnose skeletal dysplasias using radiographic images, dREAMS, is available online (registration or subscription required).
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with COMP-related pseudoachondroplasia (COMP-PSACH), the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
COMP-Related Pseudoachondroplasia: Recommended Evaluations Following Initial Diagnosis
Treatment of Manifestations
There is no cure for COMP-PSACH.
Supportive care to improve quality of life, maximize function, and reduce complications is recommended. This ideally involves multidisciplinary care by specialists in relevant fields (see Table 4).
Table 4.
COMP-Related Pseudoachondroplasia: Treatment of Manifestations
Surveillance
To monitor existing manifestations, the individual's response to supportive care, and the emergence of new manifestations, the evaluations summarized in Table 5 are recommended.
Table 5.
COMP-Related Pseudoachondroplasia: Recommended Surveillance
Agents/Circumstances to Avoid
In the small fraction of individuals with odontoid hypoplasia, extreme neck flexion and extension should be avoided.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Pregnancy Management
For females with COMP-PSACH, delivery by cesarean section is often necessary because of the small size of the pelvis. Cesarean delivery should be considered on a case-by-case basis.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Other
Growth hormone treatment is ineffective in COMP-PSACH [Kanazawa et al 2003].
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
COMP-related pseudoachondroplasia (COMP-PSACH) is inherited in an autosomal dominant manner.
Risk to Family Members
Parents of a proband
- Some individuals diagnosed with COMP-PSACH have an affected parent.
- A proband diagnosed with COMP-PSACH may have the disorder as the result of a de novo pathogenic variant. The proportion of probands who have a de novo pathogenic variant has not been accurately determined, but a study by Kennedy and colleagues indicated that in at least 22% of individuals with molecularly confirmed COMP-PSACH, a COMP pathogenic variant had arisen de novo [Kennedy et al 2005a].
- If the proband appears to be the only affected family member (i.e., a simplex case), evaluation of the parents of the proband is recommended in order to evaluate their genetic status and inform recurrence risk assessment; recommended evaluations include physical examination, radiographs, and molecular genetic testing. (Molecular genetic testing may detect parental somatic mosaicism for the COMP pathogenic variant.)
- If the pathogenic variant identified in the proband is not identified in either parent and parental identity testing has confirmed biological maternity and paternity, the following possibilities should be considered:
- The proband has a de novo pathogenic variant.
- The proband inherited a pathogenic variant from a parent with germline (or somatic and germline) mosaicism.* Note: Testing of parental leukocyte DNA may not detect all instances of somatic mosaicism and will not detect a pathogenic variant that is present in the germ (gonadal) cells only. Germline mosaicism for a COMP pathogenic variant has been reported [Hall et al 1987, Ferguson et al 1997]; the frequency is unknown.* A parent with somatic and germline mosaicism for a COMP pathogenic variant may be mildly/minimally affected.
Sibs of a proband. The risk to the sibs of the proband depends on the clinical/genetic status of the proband's parents:
- If one parent of the proband has COMP-PSACH, sibs have a 50% chance of inheriting the COMP pathogenic variant and having COMP-PSACH.
- If both parents have COMP-PSACH, sibs of the proband have a 25% chance of having average stature, a 50% chance of having COMP-PSACH, and a 25% chance of having biallelic COMP pathogenic variants and severe pseudoachondroplasia [Tariq et al 2018]. Severe pseudoachondroplasia has been reported in two individuals from a consanguineous family who were found to have homozygous pathogenic COMP variants. (Note: Other family members with heterozygous variants had a mild pseudoachondroplasia phenotype that the authors felt was more typical of multiple epiphyseal dysplasia [Tariq et al 2018].)
- If the parents are clinically unaffected, the recurrence risk to the sibs of a proband appears to be low. However, sibs of a proband with clinically unaffected parents are still presumed to be at increased risk for COMP-PSACH because of the possibility of parental germline mosaicism [Hall et al 1987, Ferguson et al 1997].
Offspring of a proband
- Each child of an individual with COMP-PSACH and a reproductive partner with normal bone growth has a 50% chance of inheriting the COMP pathogenic variant and having COMP-PSACH.
- Because many individuals with short stature select reproductive partners with short stature, offspring of individuals with COMP-PSACH may be at risk of having double heterozygosity for two dominantly inherited bone growth disorders. The phenotypes of these individuals may be distinct from those of the parents [Unger et al 2001, Flynn & Pauli 2003].
- If both partners have a dominantly inherited bone growth disorder, offspring have a 25% chance of having the maternal bone growth disorder, a 25% chance of having the paternal bone growth disorder, a 25% chance of having average stature and bone growth, and a 25% chance of having double heterozygosity for the two disorders.
Other family members. The risk to other family members depends on the status of the proband's parents: if a parent is affected, the parent's family members are at risk.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults with COMP-PSACH.
Prenatal Testing and Preimplantation Genetic Testing
Once the COMP pathogenic variant has been identified in an affected family member, prenatal and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- MedlinePlus
- Dwarf Athletic Association of America
- Dwarf Sports Association UKUnited Kingdom
- ERN BONDEuropean Reference Network on Rare Bone Diseases
- Little People of AmericaPhone: 888-LPA-2001; 714-368-3689Fax: 707-721-1896Email: info@lpaonline.org
- Little People UKUnited KingdomPhone: 07925893398Email: admin@littlepeopleuk.org
- Restricted Growth AssociationUnited Kingdom
- UCLA International Skeletal Dysplasia Registry (ISDR)Phone: 310-825-8998
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
COMP-Related Pseudoachondroplasia: Genes and Databases
Table B.
OMIM Entries for COMP-Related Pseudoachondroplasia (View All in OMIM)
Molecular Pathogenesis
Cartilage oligomeric matrix protein (COMP) is composed of an amino-terminal coiled-coil domain, four type II (EGF-like) repeats, eight consecutive type III (calmodulin-like calcium-binding) repeats, and a carboxyl-terminal globular domain. The type III motifs typically are composed of both an N- and a C-type motif, although the third and fifth type III repeats lack the N-type motif. COMP is a homopentameric adhesive glycoprotein found predominantly in the cartilage extracellular matrix [Hedbom et al 1992]. COMP is also found in tendon, ligament, and muscle. COMP is a modular, multifunctional structural protein. The type III repeats bind calcium cooperatively and the carboxyl-terminal globular domain interacts with both fibrillar (types I, II, and III) and nonfibrillar (type IX) collagens.
Pathogenic variants in the exons encoding the type III repeats of COMP result in the misfolding of the mutated protein and its retention in the rough endoplasmic reticulum (rER) of chondrocytes. This protein retention results in ER stress that ultimately causes increased cell death in vitro [Chen et al 2000, Maddox et al 2000, Unger & Hecht 2001, Kleerekoper et al 2002, Coustry et al 2012]. The retained protein in cartilage samples from individuals with COMP-related pseudoachondroplasia (COMP-PSACH) can have a diagnostic lamellar appearance by transmission electron microscopy [Maynard et al 1972].
The effect of pathogenic variants in the exons encoding the C-terminal globular domain of COMP is not fully resolved, but these pathogenic variants are not thought to prevent the secretion of mutated COMP in vitro [Spitznagel et al 2004, Schmitz et al 2006]. Furthermore, they are believed to affect collagen fibrillogenesis in cell culture models [Hansen et al 2011].
All individuals with pseudoachondroplasia appear to have COMP pathogenic variants [Jackson et al 2012]. Furthermore, all of the pathogenic variants predict an alteration in the primary structure of the protein, with the majority found in the exons encoding the eight type III calcium-binding repeats of the protein (~85%; exons 8-14). Pathogenic variants in the exons encoding the carboxyl-terminal globular domain have mostly been found in the remaining affected individuals (~15%; exons 14-19). Two variants in exons 7 and 8 encoding a type II repeat have been identified, but their pathogenesis has not been fully resolved [Jackson et al 2012, Briggs et al 2014].
Approximately 30% of individuals have the same pathogenic variant: deletion of a single aspartic acid codon p.Asp473 within a run of five consecutive GAC (Asp-encoding) codons in exon 13 [Hecht et al 1995, Briggs & Chapman 2002], corresponding to the seventh type III calcium-binding repeat of the protein. Most of the remaining individuals have a diverse range of single amino-acid substitution variants, small in-frame deletions, duplications, or indels. Interestingly, unlike the pathogenic variants in nucleotides of the type III repeats, pathogenic variants within the carboxyl-terminal domain (exons 14-19) appear to cluster in three distinct regions and affect only a limited number of residues. These variant clusters include p.Thr529Ile, p.Glu583Lys, p.Thr585Met, p.Thr585Arg, p.Thr585Lys, p.His587Arg, p.Gly719Ser, and p.Gly719Asp and point to an important role for these residues in the structure and/or function of COMP [Briggs et al 1998, Deere et al 1998, Hecht et al 1998, Deere et al 1999, Mabuchi et al 2001, Kennedy et al 2005a, Kennedy et al 2005b, Jackson et al 2012].
Evidence suggests that pathogenic variants in exons 7 and 8 encoding the type II repeats may be an uncommon cause of COMP-PSACH [Jackson et al 2012, Briggs et al 2014].
A single in-frame exon deletion and a single pathogenic variant predicting synthesis of a truncated protein have also been characterized but not analyzed in depth [Mabuchi et al 2003].
Mechanism of disease causation. Dominant-negative
Table 6.
COMP Pathogenic Variants Referenced in This GeneReview
Chapter Notes
Author History
Michael D Briggs, PhD (2013-present)
Daniel H Cohn, PhD; University of California, Los Angeles (2004-2013)
Michael J Wright, MB, ChB, MSc, FRCP (2013-present)
Revision History
- 30 November 2023 (sw) Comprehensive update posted live
- 16 August 2018 (sw) Comprehensive update posted live
- 16 July 2015 (me) Comprehensive update posted live
- 28 February 2013 (me) Comprehensive update posted live
- 13 April 2010 (me) Comprehensive update posted live
- 11 December 2006 (me) Comprehensive update posted live
- 20 August 2004 (ca) Review posted live
- 6 April 2004 (dc) Original submission
References
Literature Cited
- Andrzejewski A, Péjin Z, Finidori G, Badina A, Glorion C, Wicart P. Can Chiari osteotomy favorably influence long-term hip degradation in multiple epiphyseal dysplasia and pseudoachondroplasia? J Pediatr Orthop. 2021;41:e135-e140. [PubMed: 33165262]
- Briggs MD, Brock J, Ramsden SC, Bell PA. Genotype to phenotype correlations in cartilage oligomeric matrix protein associated chondrodysplasias. Eur J Hum Genet. 2014;22:1278–82. [PMC free article: PMC4051597] [PubMed: 24595329]
- Briggs MD, Chapman KL. Pseudoachondroplasia and multiple epiphyseal dysplasia: mutation review, molecular interactions, and genotype to phenotype correlations. Hum Mutat. 2002;19:465–78. [PubMed: 11968079]
- Briggs MD, Mortier GR, Cole WG, King LM, Golik SS, Bonaventure J, Nuytinck L, De Paepe A, Leroy JG, Biesecker L, Lipson M, Wilcox WR, Lachman RS, Rimoin DL, Knowlton RG, Cohn DH. Diverse mutations in the gene for cartilage oligomeric matrix protein in the pseudoachondroplasia-multiple epiphyseal dysplasia disease spectrum. Am J Hum Genet. 1998;62:311–9. [PMC free article: PMC1376889] [PubMed: 9463320]
- Chen H, Deere M, Hecht JT, Lawler J. Cartilage oligomeric matrix protein is a calcium-binding protein, and a mutation in its type 3 repeats causes conformational changes. J Biol Chem. 2000;275:26538–44. [PubMed: 10852928]
- Coustry F, Posey KL, Liu P, Alcorn JL, Hecht JT. D469del-COMP retention in chondrocytes stimulates caspase-independent necroptosis. Am J Pathol. 2012;180:738–48. [PMC free article: PMC3349870] [PubMed: 22154936]
- Deere M, Sanford T, Ferguson HL, Daniels K, Hecht JT. Identification of twelve mutations in cartilage oligomeric matrix protein (COMP) in patients with pseudoachondroplasia. Am J Med Genet. 1998;80:510–3. [PubMed: 9880218]
- Deere M, Sanford T, Francomano CA, Daniels K, Hecht JT. Identification of nine novel mutations in cartilage oligomeric matrix protein in patients with pseudoachondroplasia and multiple epiphyseal dysplasia. Am J Med Genet. 1999;85:486–90. [PubMed: 10405447]
- Délot E, King LM, Briggs MD, Wilcox WR, Cohn DH. Trinucleotide expansion mutations in the cartilage oligomeric matrix protein (COMP) gene. Hum Mol Genet. 1999;8:123–8. [PubMed: 9887340]
- Ferguson HL, Deere M, Evans R, Rotta J, Hall JG, Hecht JT. Mosaicism in pseudoachondroplasia. Am J Med Genet. 1997;70:287–91. [PubMed: 9188668]
- Flynn MA, Pauli RM. Double heterozygosity in bone growth disorders: four new observations and review. Am J Med Genet. 2003;121A:193–208. [PubMed: 12923858]
- Hall JG, Dorst JP, Rotta J, McKusick VA. Gonadal mosaicism in pseudoachondroplasia. Am J Med Genet. 1987;28:143–51. [PubMed: 3314506]
- Hansen U, Platz N, Becker A, Bruckner P, Paulsson M, Zaucke F. A secreted variant of cartilage oligomeric matrix protein carrying a chondrodysplasia-causing mutation (p.H587R) disrupts collagen fibrillogenesis. Arthritis Rheum. 2011;63:159–67. [PubMed: 20936634]
- Hecht JT, Montufar-Solis D, Decker G, Lawler J, Daniels K, Duke PJ. Retention of cartilage oligomeric matrix protein (COMP) and cell death in redifferentiated pseudoachondroplasia chondrocytes. Matrix Biol. 1998;17:625–33. [PubMed: 9923655]
- Hecht JT, Nelson LD, Crowder E, Wang Y, Elder FF, Harrison WR, Francomano CA, Prange CK, Lennon GG, Deere M, Lawler J. Mutations in exon 17B of cartilage oligomeric matrix protein (COMP) cause pseudoachondroplasia. Nat Genet. 1995;10:325–9. [PubMed: 7670471]
- Hedbom E, Antonsson P, Hjerpe A, Aeschlimann D, Paulsson M, Rosa-Pimentel E, Sommarin Y, Wendel M, Oldberg A, Heinegard D. Cartilage matrix proteins. An acidic oligomeric protein (COMP) detected only in cartilage. J Biol Chem. 1992;267:6132–6. [PubMed: 1556121]
- Horton WA, Hall JG, Scott CI, Pyeritz RE, Rimoin DL. Growth curves for height for diastrophic dysplasia, spondyloepiphyseal dysplasia congenita, and pseudoachondroplasia. Am J Dis Child. 1982;136:316–9. [PubMed: 6803579]
- Hunter AG. Perceptions of the outcome of orthopedic surgery in patients with chondrodysplasias. Clin Genet. 1999;56:434–40. [PubMed: 10665662]
- Jackson GC, Mittaz-Crettol L, Taylor JA, Mortier GR, Spranger J, Zabel B, Le Merrer M, Cormier-Daire V, Hall CM, Offiah A, Wright MJ, Savarirayan R, Nishimura G, SC, Elles R, Bonafe L, Superti-Furga A, Unger S, Zankl A, Briggs MD. Pseudoachondroplasia and multiple epiphyseal dysplasia: a 7-year comprehensive analysis of the known disease genes identify novel and recurrent mutations and provides an accurate assessment of their relative contribution. Hum Mutat. 2012;33:144–57. [PMC free article: PMC3272220] [PubMed: 21922596]
- Kanazawa H, Tanaka H, Inoue M, Yamanaka Y, Namba N, Seino Y. Efficacy of growth hormone therapy for patients with skeletal dysplasia. J Bone Miner Metab. 2003;21:307–10. [PubMed: 12928832]
- Kennedy J, Jackson G, Ramsden S, Taylor J, Newman W, Wright MJ, Donnai D, Elles R, Briggs MD. COMP mutation screening as an aid for the clinical diagnosis and counselling of patients with a suspected diagnosis of pseudoachondroplasia or multiple epiphyseal dysplasia. Eur J Hum Genet. 2005a;13:547–55. [PMC free article: PMC2673054] [PubMed: 15756302]
- Kennedy J, Jackson GC, Barker FS, Nundlall S, Bella J, Wright MJ, Mortier GR, Neas K, Thompson E, Elles R, Briggs MD. Novel and recurrent mutations in the C-terminal domain of COMP cluster in two distinct regions and result in a spectrum of phenotypes within the pseudoachondroplasia -- multiple epiphyseal dysplasia disease group. Hum Mutat. 2005b;25:593–4. [PubMed: 15880723]
- Kleerekoper Q, Hecht JT, Putkey JA. Disease-causing mutations in cartilage oligomeric matrix protein cause an unstructured Ca2+ binding domain. J Biol Chem. 2002;277:10581–9. [PubMed: 11782471]
- Li QW, Song HR, Mahajan RH, Suh SW, Lee SH. Deformity correction with external fixator in pseudoachondroplasia. Clin Orthop Relat Res. 2007;454:174–9. [PubMed: 16957646]
- Mabuchi A, Haga N, Ikeda T, Manabe N, Ohashi H, Takatori Y, Nakamura K, Ikegawa S. Novel mutation in exon 18 of the cartilage oligomeric matrix protein gene causes a severe pseudoachondroplasia. Am J Med Genet. 2001;104:135–9. [PubMed: 11746044]
- Mabuchi A, Manabe N, Haga N, Kitoh H, Ikeda T, Kawaji H, Tamai K, Hamada J, Nakamura S, Brunetti-Pierri N, Kimizuka M, Takatori Y, Nakamura K, Nishimura G, Ohashi H, Ikegawa S. Novel types of COMP mutations and genotype-phenotype association in pseudoachondroplasia and multiple epiphyseal dysplasia. Hum Genet. 2003;112:84–90. [PubMed: 12483304]
- Maddox BK, Mokashi A, Keene DR, Bächinger HP. A cartilage oligomeric matrix protein mutation associated with pseudoachondroplasia changes the structural and functional properties of the type 3 domain. J Biol Chem. 2000;275:11412–7. [PubMed: 10753957]
- Maynard JA, Cooper RR, Ponseti IV. A unique rough surfaced endoplasmic reticulum inclusion in pseudoachondroplasia. Lab Invest. 1972;26:40–4. [PubMed: 4333078]
- McKeand J, Rotta J, Hecht JT. Natural history study of pseudoachondroplasia. Am J Med Genet. 1996;63:406–10. [PubMed: 8725795]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24. [PMC free article: PMC4544753] [PubMed: 25741868]
- Schmitz M, Becker A, Schmitz A, Weirich C, Paulsson M, Zaucke F, Dinser R. Disruption of extracellular matrix structure may cause pseudoachondroplasia phenotypes in the absence of impaired cartilage oligomeric matrix protein secretion. J Biol Chem. 2006;281:32587–95. [PubMed: 16928687]
- Shetty GM, Song HR, Unnikrishnan R, Suh SW, Lee SH, Hur CY. Upper cervical spine instability in pseudoachondroplasia. J Pediatr Orthop. 2007;27:782–7. [PubMed: 17878785]
- Spitznagel L, Nitsche DP, Paulsson M, Maurer P, Zaucke F. Characterization of a pseudoachondroplasia-associated mutation (His587-->Arg) in the C-terminal, collagen-binding domain of cartilage oligomeric matrix protein (COMP). Biochem J. 2004;377:479–87. [PMC free article: PMC1223886] [PubMed: 14580238]
- Spranger JW, Zabel B, Kennedy J, Jackson G, Briggs M. A disorder resembling pseudoachondroplasia but without COMP mutation. Am J Med Genet A. 2005;132A:20–4. [PubMed: 15551305]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197-207. [PMC free article: PMC7497289] [PubMed: 32596782]
- Tariq M, Khan TN, Lundin L, Jameel M, Lönnerholm T, Baig SM, Dahl N, Klar J. Homozygosity for a missense variant in COMP gene associated with severe pseudoachondroplasia. Clin Genet. 2018;93:182–6. [PubMed: 28685811]
- Unger S, Ferreira CR, Mortier GR, Ali H, Bertola DR, Calder A, Cohn DH, Cormier-Daire V, Girisha KM, Hall C, Krakow D, Makitie O, Mundlos S, Nishimura G, Robertson SP, Savarirayan R, Sillence D, Simon M, Sutton VR, Warman ML, Superti-Furga A. Nosology of genetic skeletal disorders: 2023 revision. Am J Med Genet A. 2023;191:1164-209. [PMC free article: PMC10081954] [PubMed: 36779427]
- Unger S, Hecht JT. Pseudoachondroplasia and multiple epiphyseal dysplasia: New etiologic developments. Am J Med Genet. 2001;106:244–50. [PubMed: 11891674]
- Unger S, Korkko J, Krakow D, Lachman RS, Rimoin DL, Cohn DH. Double heterozygosity for pseudoachondroplasia and spondyloepiphyseal dysplasia congenita. Am J Med Genet. 2001;104:140–6. [PubMed: 11746045]
- Wynne-Davies R, Hall CM, Young ID. Pseudoachondroplasia: clinical diagnosis at different ages and comparison of autosomal dominant and recessive types. A review of 32 patients (26 kindreds). J Med Genet. 1986;23:425–34. [PMC free article: PMC1049780] [PubMed: 3783619]
- Zankl A, Jackson GC, Crettol LM, Taylor J, Elles R, Mortier GR, Spranger J, Zabel B, Unger S, Merrer ML, Cormier-Daire V, Hall CM, Wright MJ, Bonafe L, Superti-Furga A, Briggs MD. Preselection of cases through expert clinical and radiological review significantly increases mutation detection rate in multiple epiphyseal dysplasia. Eur J Hum Genet. 2007;15:150–4. [PMC free article: PMC2670452] [PubMed: 17133256]
Publication Details
Author Information and Affiliations
Newcastle University
International Centre for Life
Newcastle upon Tyne, United Kingdom
Newcastle upon Tyne Hospitals
Newcastle upon Tyne, United Kingdom
Publication History
Initial Posting: August 20, 2004; Last Update: November 30, 2023.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Briggs MD, Wright MJ. COMP-Related Pseudoachondroplasia. 2004 Aug 20 [Updated 2023 Nov 30]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.