Summary
Clinical characteristics.
SYNE1 deficiency comprises a phenotypic spectrum that ranges from autosomal recessive cerebellar ataxia at the mild end to arthrogryposis multiplex congenita (AMC) at the severe end. SYNE1-deficient cerebellar ataxia, the most commonly recognized manifestation of SYNE1 deficiency to date, is a slowly progressive disorder typically beginning in adulthood (age range 6-45 years). While some individuals have a pure cerebellar syndrome (i.e., cerebellar ataxia, dysarthria, dysmetria, abnormalities in ocular saccades and smooth pursuit), many also have upper motor neuron dysfunction (spasticity, hyperreflexia, Babinski sign) and/or lower motor neuron dysfunction (amyotrophy, reduced reflexes, fasciculations). Most individuals develop features of the cerebellar cognitive and affective syndrome (i.e., significant deficits in attention, executive functioning, verbal working memory, and visuospatial/visuoconstructional skills). The two less common phenotypes are SYNE1-deficient childhood-onset multisystem disease (ataxia, upper and lower motor neuron dysfunction, muscle weakness and wasting, intellectual disability) and SYNE1-deficient arthrogryposis multiplex congenita (decreased fetal movements and severe neonatal hypotonia associated with multiple congenital joint contractures including clubfoot).
Diagnosis/testing.
The diagnosis of SYNE1 deficiency is established in a proband with suggestive findings and biallelic SYNE1 pathogenic variants identified by molecular genetic testing.
Management.
Treatment of manifestations: There is no specific treatment for SYNE1 deficiency. The goals of treatment are to maximize function and reduce complications. Each affected individual should be managed by a multidisciplinary team of relevant specialists including neurologists, occupational therapists, physical therapists, physiatrists, orthopedists, nutritionists, speech therapists, respiratory therapists, and psychologists depending on the clinical manifestations.
Surveillance: Annual (or more often as needed) neurologic examination; assessment of mobility and self-help skills (as they relate to ataxia, spasticity, weakness), dysarthria, dysphagia, cognition, and psychiatric manifestations.
Genetic counseling.
SYNE1 deficiency is inherited in an autosomal recessive manner. The parents of an affected individual are obligate heterozygotes (i.e., carriers of one SYNE1 pathogenic variant). At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the SYNE1 pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives, prenatal diagnosis for a pregnancy at increased risk, and preimplantation genetic testing are possible.
GeneReview Scope
Table
SYNE1 cerebellar ataxia (autosomal recessive cerebellar ataxia 1 [ARCA1]) SYNE1-deficient arthrogryposis multiplex congenita (AMC)
Diagnosis
SYNE1 deficiency comprises a phenotypic spectrum that ranges from autosomal recessive cerebellar ataxia to arthrogryposis multiplex congenita (AMC).
Suggestive Findings
SYNE1 deficiency should be suspected in individuals with a combination of the following clinical features and/or clinical syndrome based on age of onset.
Clinical features
- Cerebellar ataxia
- Progressive ataxia of gait
- Clumsiness of hands
- Dysmetria
- Dysarthria
- Abnormalities in ocular saccades and smooth pursuit
- Upper and/or lower motor neuron involvement
- Spasticity, hyperactive deep tendon reflexes, extensor plantar response
- Muscle atrophy, diminished deep tendon reflexes, fasciculations
- Cognitive impairment
- Delayed motor milestones in infancy
- Intellectual disability
- Cognitive dysfunction typical of the cerebellar cognitive and affective syndrome (deficits in executive functioning, language, visuospatial/visuoconstructional skills)
- Skeletal involvement
- Scoliosis or kyphosis
- Pes cavus
- Arthrogryposis with distal joint contractures
Clinical syndrome, defined according to age at onset:
- Neonatal onset. Arthrogryposis multiplex congenita (AMC); neonatal hypotonia with decreased fetal movements resulting in distal joint contractures (including bilateral clubfoot, adducted thumbs, flexion contractures of fingers) followed by delayed motor milestones and progressive motor decline after the first decade [Attali et al 2009, Baumann et al 2017]
- Childhood onset. Multisystem phenotype; childhood-onset ataxia with upper and lower motor neuron dysfunction, elevation of serum CK concentration, pes cavus, other skeletal and soft tissue anomalies, intellectual disability, followed by respiratory insufficiency in adolescence [Synofzik & Schüle 2017]
- Adult onset. Cerebellar ataxia (ARCA1); cerebellar ataxia, frequent upper and/or lower motor neuron involvement, and cognitive impairment typical of the cerebellar cognitive and affective syndrome [Dupré et al 2007, Synofzik et al 2016]
Electrophysiologic Studies
Cerebellar ataxia
- Nerve conduction studies. Usually normal; may occasionally show an axonal neuropathy [Synofzik et al 2016, Yucesan et al 2017]
- Electromyography (EMG). Usually normal; may occasionally show acute or chronic neurogenic changes in individuals with clinical evidence of motor neuron dysfunction [Izumi et al 2013, Synofzik et al 2016]
AMC
- Nerve conduction studies and EMG (in newborns). May be normal [Baumann et al 2017]
- EMG (later onset). Includes mild chronic neurogenic findings [Attali et al 2009]
Brain Imaging
Brain MRI in individuals with childhood-onset multisystem disease or adult-onset ataxia usually shows marked diffuse cerebellar atrophy with no other abnormalities (Figure 1).
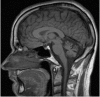
Figure 1.
MRI of a female age 29 years with SYNE1 adult-onset cerebellar ataxia. Sagittal T1-weighted imaging shows marked diffuse cerebellar atrophy with no atrophy of the cerebral cortex, midbrain, pons, or medulla.
- Brain stem atrophy has been reported in one individual with childhood-onset multisystem disease [Izumi et al 2013].
- White matter abnormalities in the brain and spinal cord that mimicked findings in multiple sclerosis have been reported in two individuals with adult-onset ataxia [Algahtani et al 2017].
18F-FDG-PET imaging shows marked homogeneous hypometabolism in the cerebellar hemispheres. Pontine brain stem hypometabolism has been reported in one individual with motor neuron involvement [Synofzik et al 2016].
Establishing the Diagnosis
The diagnosis of SYNE1 deficiency is established in a proband with suggestive findings and biallelic SYNE1 pathogenic variants identified by molecular genetic testing (see Table 1).
Because the phenotype of SYNE1 deficiency is indistinguishable from many other inherited disorders with similar complex neurologic and neuromuscular phenotypes, molecular genetic testing approaches include comprehensive genomic testing or use of a multigene panel [Dupré et al 2007, Baumann et al 2017, Coutelier et al 2018, Sun et al 2019].
Note: Single-gene testing (sequence analysis of SYNE1, followed by gene-targeted deletion/duplication analysis) is rarely useful and typically NOT recommended.
- Comprehensive genomic testing (which does not require the clinician to determine which gene[s] are likely involved) is the best option. Exome sequencing is most commonly used; genome sequencing is also possible. If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
- A multigene panel that includes SYNE1 and other genes of interest (see Differential Diagnosis) may be considered to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Table 1.
Molecular Genetic Testing Used in SYNE1 Deficiency
Clinical Characteristics
Clinical Description
The phenotype and severity of SYNE1 deficiency vary widely and span a spectrum ranging from adult-onset cerebellar ataxia at the milder end to childhood-onset multisystem disease and prenatal-onset arthrogryposis multiplex congenita at the more severe end.
SYNE1 Adult-Onset Cerebellar Ataxia
SYNE1-deficient cerebellar ataxia, also known as autosomal recessive cerebellar ataxia 1 (ARCA1), typically begins in adulthood (mean age at onset: 31.6 years [Dupré et al 2007]; range 6 to 45 years in reported series).
The initial description of ARCA1 was that of a pure cerebellar syndrome characterized by cerebellar ataxia, dysarthria, dysmetria, and abnormalities in ocular saccades and smooth pursuit [Dupré et al 2007]. In individuals from the original series, 33% had brisk reflexes and 6% had positive Babinski signs and/or ankle clonus, suggesting mild upper motor neuron involvement. Subsequently, Synofzik et al [2016] reported motor neuron dysfunction in as many as 58% of affected individuals, comprising 31% with pure upper motor neuron dysfunction (spasticity, positive bilateral Babinski signs), 19% with combined upper and lower motor neuron dysfunction, and 8% with pure lower motor neuron dysfunction (amyotrophy, reduced reflexes, fasciculations, or neurogenic changes on EMG). Slow saccades have been reported along with other oculomotor abnormalities, including square wave jerks, ophthalmoparesis, and strabismus. Skeletal involvement with scoliosis and pes cavus is an associated finding in some individuals. Reduced sense of vibration, polyneuropathy, and urge incontinence are rare occurrences [Synofzik et al 2016]. There is no evidence of muscle disease, and creatine kinase values are normal.
Individuals with SYNE1-deficient cerebellar ataxia show typical findings of the cerebellar cognitive and affective syndrome: significant deficits in attention, executive functioning, verbal working memory, and visuospatial/visuoconstructional skills [Laforce et al 2010, Mademan et al 2016].
The disease course is usually slowly progressive, resulting in a moderate degree of disability but normal life expectancy [Dupré et al 2007].
SYNE1 Childhood-Onset Multisystem Disease
A rarer phenotype is childhood-onset complex and severe multisystem disease with ataxia, upper and lower motor neuron dysfunction, pes cavus, intellectual disability, and findings suggestive of muscle disease (weakness, muscle wasting, elevated creatine kinase values, respiratory insufficiency) [Synofzik et al 2016]. Involvement of bulbar muscles may be prominent with tongue fasciculations and atrophy as well as slurred speech, and the clinical picture may mimic amyotrophic lateral sclerosis [Izumi et al 2013]. Respiratory insufficiency may present with respiratory distress or restrictive lung disease and may require noninvasive or mechanical ventilation. The reported broad range of skeletal and soft tissue abnormalities includes sacral cysts, pseudoarthrosis clavicula, hyperlaxity of joints, Achilles tendon contractures, kyphosis, scoliosis, pes cavus, cataract, and hypertelorism.
One individual had developmental abnormalities of the visceral organs (i.e., malrotation of the colon and unilateral position of both kidneys) [Mademan et al 2016, Synofzik et al 2016].
Brain MRI may show brain stem atrophy in addition to cerebellar atrophy [Izumi et al 2013].
Death between ages 36 and 44 years has been reported in a few individuals [Izumi et al 2013, Synofzik et al 2016].
SYNE1 Arthrogryposis Multiplex Congenita
SYNE1-deficient arthrogryposis multiplex congenita is characterized by decreased fetal movements in the absence of polyhydramnios, intrauterine growth restriction, or associated malformations [Attali et al 2009, Baumann et al 2017]. Neonates have severe hypotonia presenting as "floppy infant" with bilateral clubfeet, distal joint contractures with adducted thumbs and flexion contractures of fingers, and cryptorchidism in males (see Baumann et al [2017], Figure 1). Proximal weakness, facial weakness, and decreased or absent deep tendon reflexes have been reported. Motor milestones are delayed, followed by progressive motor decline after the first decade. Affected individuals use the Gower maneuver when arising from a squatting position and have limited ability to ambulate independently and to alternate their feet when climbing stairs. Flexion contractures of the proximal interphalangeal joints of the third and fourth fingers may persist despite adequate management.
Cerebellar involvement and pyramidal signs have not been reported. Intellectual development is borderline to normal. Growth deficiency worsens with advancing age despite adequate weight gain. Hyperopia with intermittent strabismus has been reported.
Early death has been reported in two individuals: one age 22 years with severe kyphoscoliosis and restrictive lung disease who died of pneumonia and sepsis [Attali et al 2009], and an infant age four months who presented with severe neonatal hypotonia and respiratory failure. Note that the infant did not undergo genetic testing but had a sib with confirmed SYNE1 deficiency [Baumann et al 2017].
In SYNE1-deficient AMC, muscle biopsy may show variations in the size of muscle fibers without increased number of muscle fibers with central nuclei. Creatine kinase values are normal.
Genotype-Phenotype Correlations
Most pathogenic variants associated with the ARCA1 phenotype are nonsense or frameshift and are localized throughout the gene, excluding the KASH domain. Most – but not all – SYNE1 pathogenic variants associated with motor neuron involvement are located toward the 3' end of the gene [Yoshinaga et al 2017].
SYNE1 pathogenic variants associated with arthrogryposis multiplex congenita (AMC) are distal truncating variants that are expected to lead to a truncated Nesprin1α (or Nesprin1α2) isoform, which is muscle and retina specific [Duong et al 2014, Potter et al 2017].
Nomenclature
In this GeneReview, the term "SYNE1 deficiency" refers to the full neurologic and neuromuscular phenotypic spectrum of biallelic SYNE1 pathogenic variants: from autosomal recessive cerebellar ataxia 1 (ARCA1) at the mild end of the continuum to SYNE1-AMC at the most severe end of the continuum.
Note: Autosomal recessive cerebellar ataxia 1 (ARCA1) has also been referred to as "recessive ataxia of Beauce" and "spinocerebellar ataxia recessive 8" (SCAR8).
Most recently, the International Parkinson and Movement Disorder Society Task Force on Classification and Nomenclature of Genetic Movement Disorders suggested the term "ATX-SYNE1" [Rossi et al 2018].
Prevalence
ARCA1, initially described in the French Canadian population, has now been reported worldwide, notably in Japan, Europe, the Middle East, and Brazil. Although its exact prevalence is not known, it is highly prevalent in the French Canadian population, which is a homogeneous founder population [Dupré et al 2007].
When Friedreich ataxia has been excluded, SYNE1 deficiency represents 5.3%-6% of unexplained early-onset (i.e., age <40 years) autosomal recessive ataxias [Mademan et al 2016, Synofzik et al 2016].
The prevalence of SYNE1-deficient arthrogryposis multiplex congenita cannot be evaluated as it has only been reported in a few families to date.
Genetically Related (Allelic) Disorders
Heterozygous SYNE1 pathogenic variants are also an uncommon cause of Emery-Dreifuss muscular dystrophy type 4 (EDMD4) or Emery-Dreifuss muscular dystrophy-like, characterized by early joint contractures, progressive muscular atrophy, and muscle weakness, with cardiomyopathy in EDMD [Zhang et al 2007] or without cardiac involvement in EDMD-like [Chen et al 2017]. Typically, affected individuals manifest the following: (1) early contractures of the Achilles tendons, elbows, and posterior cervical muscles (progression of which eventually severely limits anterior spinal flexion); (2) progressive skeletal muscle weakness and wasting; and (3) severe cardiomyopathy with ventricular and supraventricular arrhythmias, chamber dilatation, and heart failure as a consequence of myocardial cell replacement by fibrosis and adipose tissue [Chen et al 2017]. Heart transplantation may be required.
Differential Diagnosis
Table 2.
Disorders to Consider in the Differential Diagnosis of SYNE1 Deficiency
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with SYNE1 deficiency, the evaluations summarized in this section (if not performed as part of the evaluation that led to the diagnosis) are recommended:
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with SYNE1-Deficient Cerebellar Ataxia
Table 4.
Recommended Evaluations Following Initial Diagnosis in Individuals with SYNE1-Deficient Arthrogryposis Multiplex Congenita
Treatment of Manifestations
There is no specific treatment for SYNE1 deficiency. The goals of treatment are to maximize function and reduce complications. Each affected individual should be managed by a multidisciplinary team of relevant specialists such as neurologists, occupational therapists (OT), physical therapists (PT), physiatrists, orthopedists, nutritionists, speech therapists, respiratory therapists, and psychologists depending on the clinical manifestations.
Table 5.
Treatment of Manifestations in Individuals with SYNE1-Deficient Cerebellar Ataxia
Table 6.
Treatment of Manifestations in Individuals with SYNE1-Deficient Arthrogryposis Multiplex Congenita
Surveillance
There are no published surveillance guidelines for individuals with SYNE1 deficiency or for degenerative ataxias in general.
Table 7.
Recommended Surveillance for Individuals with SYNE1 Deficient Cerebellar Ataxia
Table 8.
Recommended Surveillance for Individuals with SYNE1-Deficient Arthrogryposis Multiplex Congenita
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
SYNE1 deficiency is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- The parents of an affected individual are obligate heterozygotes (i.e., carriers of one SYNE1 pathogenic variant).
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Sibs of a proband
- At conception, each sib of an affected individual has a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk of developing the disorder.
Offspring of a proband. The offspring of an individual with SYNE1 deficiency are obligate heterozygotes (carriers) for a SYNE1 pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a SYNE1 pathogenic variant.
Carrier (Heterozygote) Detection
Carrier testing for at-risk relatives requires prior identification of the SYNE1 pathogenic variants in the family.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the SYNE1 pathogenic variants have been identified in an affected family member, prenatal diagnosis for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- AMCSI: Arthrogryposis Multiplex Congenita Support, Inc.P.O. Box 6291Spartanburg SC 29304Phone: 805-55-AMCSI (1-805-552-6274)Email: bod@amcsupport.org
- Ataxia UKUnited KingdomPhone: 0800 995 6037; +44 (0) 20 7582 1444 (from abroad)Email: help@ataxia.org.uk
- euro-ATAXIA (European Federation of Hereditary Ataxias)United KingdomEmail: lporter@ataxia.org.uk
- National Ataxia FoundationPhone: 763-553-0020Fax: 763-553-0167Email: naf@ataxia.org
- NCBI Genes and Disease
- CoRDS RegistrySanford ResearchPhone: 605-312-6300
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
SYNE1 Deficiency: Genes and Databases
Table B.
OMIM Entries for SYNE1 Deficiency (View All in OMIM)
Molecular Pathogenesis
Gene structure. SYNE1 is one of the largest genes in the human genome with 0.5 Mb of genomic DNA. Alternatively spliced transcript variants encoding different protein isoforms have been described. The longest transcript NM_182961.3 (27,748 bp; variant 1) has 146 exons, of which exons 1 and 2 are noncoding. A shorter transcript variant NM_033071.3 (27,439 bp; variant 2) has 146 exons with exon 1 noncoding; this variant has multiple differences in the coding region but maintains the same reading frame as transcript variant 1.
See Table A, Gene for a detailed summary of gene, transcripts, and protein isoforms.
Pathogenic variants. Most pathogenic variants associated with the ARCA1 phenotype are nonsense or frameshift and are localized throughout the gene, excluding the KASH domain [Yoshinaga et al 2017]. Pathogenic variants associated with arthrogryposis multiplex congenita (AMC) are distal truncating variants that are expected to lead to a truncated Nesprin1α (or Nesprin1α2) isoform, which is muscle and retina specific [Duong et al 2014, Potter et al 2017].
Normal gene product. SYNE1 encodes a multi-isomeric protein called nesprin1, a scaffold protein involved in the binding of the nuclear membrane and the cytoskeleton. Nesprin-1 localizes at the outer nuclear membrane, where it interacts with SUN proteins located at the inner nuclear membrane to form the linker of the nucleoskeleton and cytoskeleton (LINC) complex [Sosa et al 2012]. The longest transcript variant NM_182961.3 encodes a 8,797-amino-acid protein (>1000 kd) known as the nesprin-1 giant isoform (Nes1g or isoform-1 or enaptin) (NP_892006.3) [Gros-Louis et al 2007]. The nesprin-1 giant protein contains two N-terminal paired calponin homology domains that bind cytoskeletal actin, a transmembrane domain, multiple spectrin repeats that mediate anchoring and interaction with other proteins and organelles, and a C-terminal KASH domain that localizes the protein to the nuclear envelope. Transcript variant NM_033071.3 encodes an 8,749-amino-acid protein known as KLNes1g (or nesprin-1 isoform 2) (NP_149062.1) [Razafsky & Hodzic 2015]. The shorter KLNes1g isoform lacks the C-terminal KASH domain.
Two isoforms of the proteins are specifically expressed in the central nervous system when compared with other isoforms and with the related Nesprin2 protein: Nes1g is particularly expressed in CNS tissues along with its KLNes1g, which is abundantly expressed in the cerebellum [Gros-Louis et al 2007, Duong et al 2014, Razafsky & Hodzic 2015].
SYNE1 has multiple alternative start and termination sites that allow for multiple isoforms lacking certain specific domains [Rajgor et al 2012; see also Razafsky & Hodzic 2015, Yoshinaga et al 2017, Potter et al 2018, and references therein]. These isoforms are present in multiple subcellular locations beyond the nuclear envelope and serve to link these structures to the actin skeleton [Zhang et al 2007]. Specific isoforms appear to have a tissue-specific transcription, and this transcription is highly adaptable according to cell needs for maintaining homeostasis [Rajgor et al 2012].
Abnormal gene product. Most pathogenic variants associated with the ARCA1 phenotype are nonsense or frameshift and are localized throughout the gene, excluding the KASH domain [Yoshinaga et al 2017]. Hence, these pathogenic variants are expected to affect the structure of the KLNes1g isoform, whose loss of function is thought to lead to ARCA1 [Razafsky & Hodzic 2015]. Indeed, the KLNes1g isoform is abundantly expressed in the cerebellum, where it localizes to essential synapses between mossy fibers and cerebellar granule neurons within the granule cell layer [Potter et al 2018]. Analyses in murine models suggest that this isoform is involved in vesicular trafficking and dendritic membrane structural organization, which indicates that defective synaptic transmission may underlie ARCA1 pathology; however, this remains to be confirmed [Razafsky & Hodzic 2015]. Pathogenic variants that alter the more C-terminal region of the protein have the potential to also alter the ubiquitously expressed Nesprin1β isoform, which may underlie the more complex multisystem phenotype observed in some individuals [Potter et al 2018].
Pathogenic variants associated with arthrogryposis multiplex congenita (AMC) are distal truncating variants that are expected to lead to a truncated Nesprin1α (or Nesprin1α2) isoform, which is muscle and retina specific [Duong et al 2014, Potter et al 2017]. There is mounting evidence that Nesprin1α is involved in skeletal muscle function: Nesprin1α is upregulated during myogenic differentiation and is required for the recruitment of centrosomal proteins to the nuclear envelope, which ensures proper nuclear positioning [Gimpel et al 2017]. Aberrant nuclear positioning is associated with other muscular diseases, suggesting that proper nuclear localization is essential for skeletal muscle function [Stroud et al 2017]. In murine studies of different Nesprin1 isoforms, loss of Nesprin1α2 led to severe nuclear mispositioning and postnatal lethality, suggesting that this is the isoform essential for skeletal muscle function [Stroud et al 2017]. Hence, loss of the Nesprin1α isoform may underlie muscular involvement in SYNE1 deficiency.
All SYNE1 pathogenic variants would also affect the Nesprin1 giant (Nes1g) isoform, which is predominantly expressed in the central nervous system at the nuclear envelope of Bergmann glia and at ciliary rootlets of ependymal cells [Baumann et al 2017, Potter et al 2018]. Mouse models with loss of Nes1g did not show any cerebellar phenotype but presented ventricular enlargement potentially reflecting cilia dysfunction [Potter et al 2018]. Of interest, scoliosis, respiratory insufficiency, and cognitive impairment are all clinical findings that have been reported in association with other ciliopathies, suggesting that loss of the Nes1g isoform may underlie some non-cerebellar and non-muscular features of the phenotype [Potter et al 2018].
References
Literature Cited
- Algahtani H, Marzouk Y, Algahtani R, Salman S, Shirah B. Autosomal recessive cerebellar ataxia type 1 mimicking multiple sclerosis: a report of two siblings with a novel mutation in SYNE1 gene in a Saudi family. J Neurol Sci. 2017;372:97–100. [PubMed: 28017257]
- Attali R, Warwar N, Israel A, Gurt I, McNally E, Puckelwartz M, Glick B, Nevo Y, Ben-Neriah Z, Melki J. Mutation of SYNE-1, encoding an essential component of the nuclear lamina, is responsible for autosomal recessive arthrogryposis. Hum Mol Genet. 2009;18:3462–9. [PubMed: 19542096]
- Baumann M, Steichen-Gersdorf E, Krabichler B, Petersen BS, Weber U, Schmidt WM, Zschocke J, Müller T, Bittner RE, Janecke AR. Homozygous SYNE1 mutation causes congenital onset of muscular weakness with distal arthrogryposis: a genotype-phenotype correlation. Eur J Hum Genet. 2017;25:262–6. [PMC free article: PMC5255944] [PubMed: 27782104]
- Bürk K, Sival DA. Scales for the clinical evaluation of cerebellar disorders. Handb Clin Neurol. 2018;154:329–39. [PubMed: 29903450]
- Chen Z, Ren Z, Mei W, Ma Q, Shi Y, Zhang Y, Li S, Xiang L, Zhang J. A novel SYNE1 gene mutation in a Chinese family of Emery-Dreifuss muscular dystrophy-like. BMC Med Genet. 2017;18:63. [PMC free article: PMC5460548] [PubMed: 28583108]
- Coutelier M, Hammer MB, Stevanin G, Monin ML, Davoine CS, Mochel F, Labauge P, Ewenczyk C, Ding J, Gibbs JR, Hannequin D, Melki J, Toutain A, Laugel V, Forlani S, Charles P, Broussolle E, Thobois S, Afenjar A, Anheim M, Calvas P, Castelnovo G, de Broucker T, Vidailhet M, Moulignier A, Ghnassia RT, Tallaksen C, Mignot C, Goizet C, Le Ber I, Ollagnon-Roman E, Pouget J, Brice A, Singleton A, Durr A, et al. Efficacy of exome-targeted capture sequencing to detect mutations in known cerebellar ataxia genes. JAMA Neurol. 2018;75:591–9. [PMC free article: PMC5885259] [PubMed: 29482223]
- Duong NT, Morris GE, Lam LT, Zhang Q, Sewry CA, Shanahan CM, Holt I. Nesprins: tissue-specific expression of epsilon and other short isoforms. PLoS One. 2014;9:e94380. [PMC free article: PMC3981789] [PubMed: 24718612]
- Dupré N, Gros-Louis F, Chrestian N, Verreault S, Brunet D, de Verteuil D, Brais B, Bouchard JP, Rouleau GA. Clinical and genetic study of autosomal recessive cerebellar ataxia type 1. Ann Neurol. 2007;62:93–8. [PubMed: 17503513]
- Gimpel P, Lee YL, Sobota RM, Calvi A, Koullourou V, Patel R, Mamchaoui K, Nédélec F, Shackleton S, Schmoranzer J, Burke B, Cadot B, Gomes ER. Nesprin-1alpha-dependent microtubule nucleation from the nuclear envelope via Akap450 is necessary for nuclear positioning in muscle cells. Curr Biol. 2017;27:2999–3009 e9. [PMC free article: PMC5640514] [PubMed: 28966089]
- Gros-Louis F, Dupré N, Dion P, Fox MA, Laurent S, Verreault S, Sanes JR, Bouchard JP, Rouleau GA. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet. 2007;39:80–5. [PubMed: 17159980]
- Haliloglu G, Topaloglu H. Arthrogryposis and fetal hypomobility syndrome. Handb Clin Neurol. 2013;113:1311–9. [PubMed: 23622356]
- Hoche F, Guell X, Vangel MG, Sherman JC, Schmahmann JD. The cerebellar cognitive affective/Schmahmann syndrome scale. Brain. 2018;141:248–70. [PMC free article: PMC5837248] [PubMed: 29206893]
- Izumi Y, Miyamoto R, Morino H, Yoshizawa A, Nishinaka K, Udaka F, Kameyama M, Maruyama H, Kawakami H. Cerebellar ataxia with SYNE1 mutation accompanying motor neuron disease. Neurology. 2013;80:600–1. [PubMed: 23325900]
- Laforce R Jr, Buteau JP, Bouchard JP, Rouleau GA, Bouchard RW, Dupré N. Cognitive impairment in ARCA-1, a newly discovered pure cerebellar ataxia syndrome. Cerebellum. 2010;9:443–53. [PubMed: 20559786]
- Mademan I, Harmuth F, Giordano I, Timmann D, Magri S, Deconinck T, Claaßen J, Jokisch D, Genc G, Di Bella D, Romito S, Schüle R, Züchner S, Taroni F, Klockgether T, Schöls L, De Jonghe P, Bauer P, Consortium E, Baets J, Synofzik M. Multisystemic SYNE1 ataxia: confirming the high frequency and extending the mutational and phenotypic spectrum. Brain. 2016;139(Pt 8):e46. [PMC free article: PMC4958896] [PubMed: 27197992]
- Martineau L, Noreau A, Dupré N. Therapies for ataxias. Curr Treat Options Neurol. 2014;16(7):300. [PubMed: 24832479]
- Potter C, Razafsky D, Wozniak D, Casey M, Penrose S, Ge X, Mahjoub MR, Hodzic D. The KASH-containing isoform of Nesprin1 giant associates with ciliary rootlets of ependymal cells. Neurobiol Dis. 2018;115:82–91. [PMC free article: PMC5943178] [PubMed: 29630990]
- Potter C, Zhu W, Razafsky D, Ruzycki P, Kolesnikov AV, Doggett T, Kefalov VJ, Betleja E, Mahjoub MR, Hodzic D. Multiple isoforms of Nesprin1 are integral components of ciliary rootlets. Curr Biol. 2017;27:2014–22 e6. [PMC free article: PMC5546243] [PubMed: 28625779]
- Rajgor D, Mellad JA, Autore F, Zhang Q, Shanahan CM. Multiple novel nesprin-1 and nesprin-2 variants act as versatile tissue-specific intracellular scaffolds. PLoS One. 2012;7:e40098. [PMC free article: PMC3388047] [PubMed: 22768332]
- Razafsky D, Hodzic D. A variant of Nesprin1 giant devoid of KASH domain underlies the molecular etiology of autosomal recessive cerebellar ataxia type I. Neurobiol Dis. 2015;78:57–67. [PMC free article: PMC4426048] [PubMed: 25843669]
- Rossi M, Anheim M, Durr A, Klein C, Koenig M, Synofzik M, Marras C, van de Warrenburg BP, et al. The genetic nomenclature of recessive cerebellar ataxias. Mov Disord. 2018;33:1056–76. [PubMed: 29756227]
- Ruffieux N, Colombo F, Gentaz E, Annoni JM, Chouiter L, Roulin Hefti S, Ruffieux A, Bihl T. Successful neuropsychological rehabilitation in a patient with cerebellar cognitive affective syndrome. Appl Neuropsychol Child. 2017;6:180–8. [PubMed: 27049666]
- Sosa BA, Rothballer A, Kutay U, Schwartz TU. LINC complexes form by binding of three KASH peptides to domain interfaces of trimeric SUN proteins. Cell. 2012;149:1035–47. [PMC free article: PMC3383001] [PubMed: 22632968]
- Stroud MJ, Feng W, Zhang J, Veevers J, Fang X, Gerace L, Chen J. Nesprin 1alpha2 is essential for mouse postnatal viability and nuclear positioning in skeletal muscle. J Cell Biol. 2017;216:1915–24. [PMC free article: PMC5496623] [PubMed: 28533284]
- Sun M, Johnson AK, Nelakuditi V, Guidugli L, Fischer D, Arndt K, Ma L, Sandford E, Shakkottai V, Boycott K, Chardon JW, Li Z, Del Gaudio D, Burmeister M, Gomez CM, Waggoner DJ, Das S. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet Med. 2019;21:195–206. [PMC free article: PMC6524765] [PubMed: 29915382]
- Synofzik M, Schüle R. Overcoming the divide between ataxias and spastic paraplegias: shared phenotypes, genes, and pathways. Mov Disord. 2017;32:332–45. [PMC free article: PMC6287914] [PubMed: 28195350]
- Synofzik M, Smets K, Mallaret M, Di Bella D, Gallenmuller C, Baets J, Schulze M, Magri S, Sarto E, Mustafa M, Deconinck T, Haack T, Züchner S, Gonzalez M, Timmann D, Stendel C, Klopstock T, Durr A, Tranchant C, Sturm M, Hamza W, Nanetti L, Mariotti C, Koenig M, Schöls L, Schüle R, de Jonghe P, Anheim M, Taroni F, Bauer P. SYNE1 ataxia is a common recessive ataxia with major non-cerebellar features: a large multi-centre study. Brain. 2016;139:1378–93. [PMC free article: PMC6363274] [PubMed: 27086870]
- van de Warrenburg BP, van Gaalen J, Boesch S, Burgunder JM, Durr A, Giunti P, Klockgether T, Mariotti C, Pandolfo M, Riess O. EFNS/ENS consensus on the diagnosis and management of chronic ataxias in adulthood. Eur J Neurol. 2014;21:552–62. [PubMed: 24418350]
- Yoshinaga T, Nakamura K, Ishikawa M, Yamaguchi T, Takano K, Wakui K, Kosho T, Yoshida K, Fukushima Y, Sekijima Y. A novel frameshift mutation of SYNE1 in a Japanese family with autosomal recessive cerebellar ataxia type 8. Hum Genome Var. 2017;4:17052. [PMC free article: PMC5656760] [PubMed: 29081981]
- Yucesan E, Ugur Iseri SA, Bilgic B, Gormez Z, Bakir Gungor B, Sarac A, Ozdemir O, Sagiroglu M, Gurvit H, Hanagasi H, Ozbek U. SYNE1 related cerebellar ataxia presents with variable phenotypes in a consanguineous family from Turkey. Neurol Sci. 2017;38:2203–7. [PubMed: 28687974]
- Zesiewicz TA, Wilmot G, Kuo SH, Perlman S, Greenstein PE, Ying SH, Ashizawa T, Subramony SH, Schmahmann JD, Figueroa KP, Mizusawa H, Schöls L, Shaw JD, Dubinsky RM, Armstrong MJ, Gronseth GS, Sullivan KL. Comprehensive systematic review summary: treatment of cerebellar motor dysfunction and ataxia: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology. 2018;90:464–71. [PMC free article: PMC5863491] [PubMed: 29440566]
- Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A, Ragnauth CD, Yi Q, Mellad JA, Warren DT, Wheeler MA, Ellis JA, Skepper JN, Vorgerd M, Schlotter-Weigel B, Weissberg PL, Roberts RG, Wehnert M, Shanahan CM. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. 2007;16:2816–33. [PubMed: 17761684]
Chapter Notes
Author History
Marie Beaudin, MD (2018-present)
Jean-Pierre Bouchard, MD; Laval University (2007-2018)
Nicolas Dupré, MD, MSc (2007-present)
Pierre-Luc Gamache, MD, PhD (2018-present)
François Gros-Louis, PhD (2007-present)
Anne Noreau, MD, PhD; University of Montreal (2011-2018)
Guy A Rouleau, MD, PhD; University of Montreal (2007-2018)
Revision History
- 6 December 2018 (bp) Comprehensive update posted live
- 13 October 2011 (cd) Revision: cognitive changes associated with this disorder as reported by Laforce et al [2010]
- 18 August 2011 (cd) Revision: mutation scanning of select exons and prenatal testing clinically available
- 14 July 2011 (me) Comprehensive update posted live
- 15 September 2009 (cd) Revision: sequence analysis and targeted mutation analysis available clinically
- 23 February 2007 (me) Review posted live
- 6 February 2007 (nd) Original submission
Publication Details
Author Information and Affiliations
Publication History
Initial Posting: February 23, 2007; Last Update: December 6, 2018.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Beaudin M, Gamache PL, Gros-Louis F, et al. SYNE1 Deficiency. 2007 Feb 23 [Updated 2018 Dec 6]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
