Summary
Clinical characteristics.
GNE myopathy is a slowly progressive muscle disease that typically presents between age 20 and 40 years with bilateral foot drop caused by anterior tibialis weakness. Lower-extremity muscle involvement progresses from the anterior to the posterior compartment of the lower leg, followed by hamstrings, then hip girdle muscles, with relative sparing of the quadriceps. A wheelchair may be needed about ten to 20 years after the onset of manifestations. The upper extremities, which may be affected within five to ten years of disease onset, do not necessarily follow a distal-to-proximal progression. In advanced stages, neck and core muscles can become affected.
Diagnosis/testing.
The diagnosis of GNE myopathy is suspected in a proband with suggestive clinical findings and muscle histopathology (rimmed vacuoles, no inflammation) and is established by the presence of biallelic pathogenic variants in GNE identified by molecular genetic testing.
Management.
Treatment of manifestations: Evaluation and management are often by a multidisciplinary team that includes neuromuscular specialists, physiatrists, and physical and occupational therapists to address issues secondary to muscle weakness, including the use of assistive ambulatory devices (e.g., ankle-foot orthoses, cane, walker, wheelchair, or powerchair). Adaptive devices to support fine motor function and activities of daily living are needed in advanced stages of the disease. Recommended evaluations also include baseline echocardiogram and pulmonary function tests in nonambulatory individuals, with management by pulmonologists as clinically indicated.
Surveillance: Follow up at least annually by neuromuscular specialists, physiatrists, and physical and occupational therapists to evaluate disease progression and address muscle strength, mobility, function, and activities of daily living; by pulmonologists to monitor respiratory muscle function in patients with advanced disease.
Agents/circumstances to avoid: Cautious use of medications/drugs with potential myotoxicity (e.g., colchicine and statins); avoidance of weight-lifting and repetitive activities that cause muscle pain.
Genetic counseling.
GNE myopathy is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a GNE pathogenic variant, each sib of an affected individual has at conception a 25% chance of inheriting both pathogenic variants and being affected, a 50% chance of inheriting one pathogenic variant and being an unaffected carrier, and a 25% chance of inheriting both normal alleles. When the GNE pathogenic variants have been identified in an affected family member, molecular genetic carrier testing of at-risk relatives, prenatal testing for pregnancies at increased risk, and preimplantation genetic testing are possible.
Diagnosis
Suggestive Findings
GNE myopathy should be suspected in individuals with the following findings.
Clinical findings
- Myopathy presenting in young adults with bilateral foot drop caused by anterior tibialis weakness, followed by slowly progressive skeletal muscle weakness. Although there is relative sparing of the quadriceps, they may become affected at late stages of the disease. The clinical picture varies depending on the stage of disease progression at which individuals are evaluated (see Clinical Description).
- Serum CK may be normal or up to four times the upper limit of normal.
Muscle histopathology
- Cryosections of affected muscles show atrophy, variation of fiber size, rimmed vacuoles, and no inflammation. The most prominent finding, the presence of rimmed vacuoles, is best identified in cryosections using modified Gomori trichrome stain and may be missed in paraffin-embedded tissue or hematoxylin and eosin staining. The "rimmed vacuoles" observed on electron microscopy that correspond to autophagic vacuoles are seen in a variety of myopathies with other etiologies that lead to autophagic degeneration (see Differential Diagnosis).
- Note: (1) Because histopathologic findings may be difficult to identify in biopsies of muscles that are unaffected or that have been replaced by fibro-fatty tissue, muscle strength or muscle MRI may aid in the identification of suitable muscles to biopsy. (2) Muscle biopsy and histopathologic examination may not be required to suspect or establish the diagnosis of GNE myopathy but remain necessary when variants of unknown significance are identified on molecular genetic testing.
Establishing the Diagnosis
The diagnosis of GNE myopathy is established in a proband with suggestive clinical findings, muscle histopathology (if performed), and biallelic pathogenic (or likely pathogenic) variants in GNE identified by molecular genetic testing (see Table 1).
Note: (1) Per ACMG variant interpretation guidelines, the terms "pathogenic variants" and "likely pathogenic variants" are synonymous in a clinical setting, meaning that both are considered diagnostic and both can be used for clinical decision making. Reference to "pathogenic variants" in this section is understood to include any likely pathogenic variants. (2) Identification of biallelic GNE variants of uncertain significance (or identification of one known GNE pathogenic variant and one GNE variant of uncertain significance) does not establish or rule out a diagnosis of GNE myopathy.
Molecular genetic testing approaches can include gene-targeted testing (single-gene testing in probands with suggestive findings or affected sibs; multigene neuromuscular panel in probands with myopathy but unspecific findings) (see Option 1). In some instances, comprehensive genomic testing (exome sequencing or genome sequencing) is performed (see Option 2).
Option 1
Single-gene testing. Sequence analysis of GNE is performed first to detect small intragenic deletions/insertions and missense, nonsense, and splice site variants. Note: Depending on the sequencing method used, single-exon, multiexon, or whole-gene deletions/duplications may not be detected. If only one or no variant is detected by the sequencing method used, the next step is to perform gene-targeted deletion/duplication analysis to detect exon and whole-gene deletions or duplications.
Note: Targeted analysis for founder pathogenic variants identified in several populations may be appropriate in some circumstances (for more details see Table 6).
- p.Met743Thr (c.2228T>C) is a founder variant in individuals of Middle Eastern ancestry [Eisenberg et al 2001, Argov et al 2003].
- p.Asp207Val (c.620A>T) and p.Val603Leu (c.1807G>C), founder variants in individuals of Asian ancestry, account for approximately 70% of disease variants in the Japanese population [Nishino et al 2002, Cho et al 2014].
A multigene panel that includes GNE and other genes of interest (see Differential Diagnosis) is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests. For this disorder a multigene panel that also includes deletion/duplication analysis is recommended (see Table 1).
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Option 2
Comprehensive genomic testing does not require the clinician to determine which gene(s) are likely involved. Exome sequencing is most commonly used; genome sequencing is also possible.
If exome sequencing is not diagnostic, exome array (when clinically available) may be considered to detect (multi)exon deletions or duplications that cannot be detected by sequence analysis.
For an introduction to comprehensive genomic testing click here. More detailed information for clinicians ordering genomic testing can be found here.
Table 1.
Molecular Genetic Testing Used in GNE Myopathy
Clinical Characteristics
Clinical Description
GNE myopathy is characterized by adult-onset slowly progressive myopathy typically presenting with bilateral foot drop, followed by distal-to-proximal lower-extremity weakness. The upper extremities, which are affected within five to ten years of disease onset, do not necessarily follow a distal-to-proximal progression, in contrast to the lower extremities. In advanced stages, neck and core muscles can also become affected.
Onset. GNE myopathy typically presents in individuals age 20-40 years with foot drop caused by anterior tibialis weakness. Rarely, in case of muscle overuse, other muscles may be affected first [de Dios et al 2014].
Progression. In the lower extremities, the disease progresses to involve muscles from the anterior compartment of the lower leg, followed by calf muscles and hamstrings, followed by hip girdle muscles, with relative sparing of the quadriceps [Argov & Yarom 1984]. The involvement of the quadriceps muscles may become evident in late stages of the disease with the rectus femoris affected first and the vastus lateralis affected last [Huizing et al 2001, Tasca et al 2012, Carrillo et al 2018].
In the upper extremities, shoulder abduction may be affected early in the disease course before grip and hand muscles are affected.
Clinical findings depend on the stage of disease progression at the time of evaluation [Quintana et al 2019]:
- Disease onset. Young adults describe symptoms such as tripping and changes in gait. On exam, there is bilateral foot drop and inability to stand on the toes or walk on the heels.
- Within five years of onset. Complete loss of ankle dorsiflexion strength, decreased knee flexion and shoulder abduction strength. Manifestations include steppage gait, some difficulty climbing stairs, and decreased balance, requiring the use of ankle-foot orthoses (AFOs).
- Five to ten years after onset. Complete loss of knee flexion strength; decreased shoulder abduction, forearm, wrist, and hand strength; quadriceps are unaffected. Manifestations include worsening gait, increased risk of falls, and poor balance requiring the use of assistive walking devices; difficulty moving from a sitting position to a standing position; and significant difficulty climbing stairs. Upper extremities: difficulty performing tasks that involve raising arms above head and initial difficulty with hand function.
- Ten to 20 years after onset. Decreased strength of hip extensors; quadriceps may be affected; the use of a wheelchair may be needed; significant difficulty with shoulder abduction and fine motor (i.e., hand) tasks. Increasing dependence for assistance with activities of daily living.
- In advanced stages. The neck, core, and respiratory muscles can be affected.
Ultimately, disease progression may result in complete loss of skeletal muscle function and dependence on caregivers. Life span is not reduced.
Other
- Respiratory muscle involvement resulting in decreased forced vital capacity has been described in the late stages of the disease; however, clinically significant involvement is rare and typically limited to individuals who are wheelchair dependent [Mori-Yoshimura et al 2013].
- Cardiac muscle is typically not affected. While cardiac involvement has been reported in persons with GNE myopathy [Chai et al 2011, Chamova et al 2015], it remains unclear whether this was due to GNE myopathy or of a different etiology.
Laboratory findings
- Serum creatine kinase (CK) activity may be normal or elevated; it typically does not exceed four times the normal value.
- Creatinine values decrease over time due to decreased muscle mass; hence, cystatin C should be used instead of creatinine to evaluate renal function.
- Mild elevation of alanine aminotransferase and aspartate aminotransferase is seen in some individuals, especially those with elevated CK.
Electromyogram and nerve conduction velocity are invasive and do not help with the diagnosis.
Muscle MRI shows fibro-fatty replacement on T1-weighted images; short tau inversion recovery hyperintensity indicates active disease.
Genotype-Phenotype Correlations
Because reports of GNE myopathy consist mainly of single individuals or relatively small series, correlations between genotype and phenotype are difficult.
Penetrance
Penetrance of biallelic GNE pathogenic variants is likely close to 100%. Only two older individuals with biallelic GNE pathogenic variants have been reported to be asymptomatic: One (age 67 years) was homozygous for the common Middle Eastern variant p.Met743Thr [Argov et al 2003] and one (age 60 years) was homozygous for the common Japanese variant p.Asp207Val [Nishino et al 2002].
Nomenclature
In order to unify the nomenclature and avoid confusion with unrelated but similarly named disorders, an international consortium proposed the term "GNE myopathy" to replace historically used terms [Huizing et al 2014].
- The phenotype was first described by Nonaka et al [1981] in Japan; at that time, the disorder was referred to as "distal myopathy with rimmed vacuoles (DMRV)" or Nonaka myopathy.
- The terms "quadriceps-sparing myopathy" and "hereditary rimmed vacuole myopathy (HIBM)" were used by Argov & Yarom [1984] and Sadeh et al [1993] to describe the GNE myopathy phenotype in affected individuals of Iranian Jewish ancestry.
With the identification of the causative gene, GNE [Eisenberg et al 2001], it became apparent that HIBM was the same disease as DMRV [Nishino et al 2002].
Prevalence
The prevalence of GNE myopathy is estimated at 1-9:1,000,000.
To date, more than 1,000 individuals with GNE myopathy and about 255 GNE variants have been reported worldwide.
The worldwide carrier rate of a pathogenic GNE variant is estimated at 1:203 individuals.
Genetically Related (Allelic) Disorders
Sialuria (OMIM 269921) is caused by heterozygous pathogenic variants in GNE. The sialuria-associated GNE variants involve the allosteric site of the epimerase enzyme activity and abolish the feedback inhibition mechanism by CMP-sialic acid, resulting in overproduction of sialic acid [Seppala et al 1999]. Affected individuals exhibit variable degrees of developmental delay, coarse facial features, and hepatomegaly. Inheritance is autosomal dominant.
Congenital thrombocytopenia has been described in individuals with biallelic GNE variants [Futterer et al 2018].
Differential Diagnosis
The differential diagnosis includes adult-onset distal myopathies and myopathies with rimmed vacuoles (see Table 2).
Table 2.
Genes of Interest in the Differential Diagnosis of GNE Myopathy
Management
Individuals with GNE myopathy are often evaluated and managed by a multidisciplinary team that includes clinical geneticists, neuromuscular specialists, physiatrists, physical and occupational therapists, and if needed, pulmonologists.
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs in an individual diagnosed with GNE myopathy, the evaluations summarized in Table 3 (if not performed as part of the evaluation that led to the diagnosis) are recommended.
Table 3.
Recommended Evaluations Following Initial Diagnosis in Individuals with GNE Myopathy
Treatment of Manifestations
There is no approved therapy for GNE myopathy. Treatment is symptomatic only (see Table 4).
Table 4.
Treatment of Manifestations in Individuals with GNE Myopathy
Surveillance
Routine follow up with the multidisciplinary team is recommended annually, or more frequently as determined by managing physician (see Table 5).
Table 5.
Recommended Multidisciplinary Team Surveillance for Individuals with GNE Myopathy
Agents/Circumstances to Avoid
It may be prudent to use medications/drugs with potential myotoxicity (e.g., colchicine, statins) with caution.
It is strongly recommended that affected individuals have a healthy diet and exercise to avoid developing hypercholesterolemia, in an effort to reduce the risk associated with taking statins.
Individuals with GNE myopathy should avoid lifting weights and performing repetitive activities that result in muscle pain.
Evaluation of Relatives at Risk
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
N-acetylmanossamine (ManNAc) is the only therapy currently in clinical development for GNE myopathy [Galeano et al 2007, Malicdan et al 2009, Niethamer et al 2012, Xu et al 2017].
Ultragenyx discontinued the clinical development of extended-release sialic acid (Ace-ER) in 2017 following a Phase III trial that failed to detect clinical efficacy [Lochmüller et al 2019].
Preclinical studies are ongoing to advance gene therapy as a potential therapy for GNE myopathy.
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members; it is not meant to address all personal, cultural, or ethical issues that may arise or to substitute for consultation with a genetics professional. —ED.
Mode of Inheritance
GNE myopathy is inherited in an autosomal recessive manner.
Risk to Family Members
Parents of a proband
- Parents of an affected individual are obligate heterozygotes (i.e., carriers for one GNE pathogenic variant).
- Molecular genetic testing is recommended for the parents of a proband to confirm that both parents are heterozygous for a GNE pathogenic variant and to allow reliable recurrence risk assessment. Although a de novo pathogenic variant has not been reported in GNE myopathy to date, de novo variants are known to occur at a low but appreciable rate in autosomal recessive disorders [Jónsson et al 2017].
- Heterozygotes (carriers) are asymptomatic and are not at risk for GNE myopathy.
Sibs of a proband
- If both parents are known to be heterozygous for a GNE pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier.
- Heterozygotes (carriers) are asymptomatic and are not at risk for GNE myopathy.
Offspring of a proband. The offspring of an individual with GNE myopathy are obligate heterozygotes (carriers) for a pathogenic variant in GNE.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of a GNE pathogenic variant.
Carrier (Heterozygote) Detection
Carrier testing for at-risk relatives requires prior identification of the GNE pathogenic variants in the family.
Related Genetic Counseling Issues
Family planning
- The optimal time for determination of genetic risk, clarification of carrier status, and discussion of the availability of prenatal/preimplantation genetic testing is before pregnancy.
- It is appropriate to offer genetic counseling (including discussion of potential risks to offspring and reproductive options) to young adults who are affected, are carriers, or are at risk of being carriers.
Prenatal Testing and Preimplantation Genetic Testing
Once the GNE pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella support organizations and/or registries for the benefit of individuals with this disorder and their families. GeneReviews is not responsible for the information provided by other organizations. For information on selection criteria, click here.
- Advancement of Research for Myopathies (ARM)P.O. Box 261926Encino 91426-1926Phone: 800-ARM-2000
- GNE Myopathy InternationalPhone: 91-9810881439Email: gne.myopathy@gmail.com
- Neuromuscular Disease Foundation (NDF)Phone: 310-721-1605; 516-441-7126
- TREAT-NMDNeuromuscular NetworkEmail: info@treat-nmd.eu
- Muscular Dystrophy Association (MDA) - USAPhone: 833-275-6321
- Muscular Dystrophy UKUnited KingdomPhone: 0800 652 6352
- Patient Association for Distal Myopathies (PADM)Email: yuriko.oda@npopadm.com
- Remudy - Registry of Muscular DystrophiesJapan
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
GNE Myopathy: Genes and Databases
Table B.
OMIM Entries for GNE Myopathy (View All in OMIM)
Molecular Pathogenesis
GNE encodes UDP-N-acetylglucosamine (GlcNAc) 2-epimerase/N-acetylmannosamine (ManNAc) kinase (GNE), a key enzyme in the intracellular biosynthesis of N-acetylneuraminic acid (Neu5Ac) [Keppler 1999] (see Figure 1). The downstream product, CMP-Neu5Ac, serves as a donor of Neu5Ac (a form of sialic acid) to glycoproteins in the Golgi which are then secreted or become plasma membrane proteins [Wopereis et al 2006, Cohen & Varki 2010].
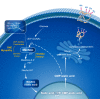
Heterozygous pathogenic variants in the allosteric site of GNE cause sialuria, an entity distinct from GNE myopathy (see Genetically Related Disorders) (OMIM 269921).
Mechanism of disease causation. Most pathogenic variants that cause GNE myopathy are missense with several shown to result in decreased epimerase and kinase enzymatic activity [Effertz et al 1999, Nishino et al 2002, Noguchi et al 2004, Sparks et al 2005, Penner et al 2006, Kurochkina et al 2010, Celeste et al 2014, Carrillo et al 2018]. No individuals homozygous for nonsense variants have been reported [Celeste et al 2014, Carrillo et al 2018]. Residual GNE enzyme activity is essential for life; complete lack of enzyme activity as demonstrated by inactivation of Gne in mice is lethal in early embryonic stages [Schwarzkopf et al 2002].
The mechanism by which disruption of GNE enzyme function causes muscle atrophy in GNE myopathy remains unclear; however, evidence suggests that the mechanism involves decreased sialylation of muscle glycoproteins. Muscle biopsies from affected individuals reveal hyposialylation of muscle glycoproteins [Noguchi et al 2004, Tajima et al 2005, Gagiannis et al 2007, Leoyklang et al 2018] including specific skeletal muscle glycoproteins, such as alpha-dystroglycan [Huizing et al 2004], NCAM [Ricci et al 2006], and neprilysin [Broccolini et al 2008].
GNE-specific laboratory technical considerations. Well-established functional studies have reported GNE enzyme activity in cells (lymphoblasts, fibroblasts) of affected individuals [Nishino et al 2002, Noguchi et al 2004, Sparks et al 2005, Celeste et al 2014].
Table 6.
Notable GNE Pathogenic Variants
Chapter Notes
Author Notes
George Karpati, MD, one of the original authors of this GeneReview, was a distinguished physician and scientist. A Hungarian-born Holocaust survivor, he became a leading expert in muscular dystrophy and other neuromuscular disorders; he held the IW Killam Chair and was Professor of Neurology and Neurosurgery at McGill University. Dr Karpati died suddenly on February 6, 2009. He leaves behind family and many close friends in Canada, the United States, Israel, and Hungary.
Acknowledgments
This study was supported Intramural Program of the National Human Genome Research Institute of the National Institutes of Health, Bethesda, Maryland, USA.
Author History
Nuria Carrillo, MD (2020-present)
Marjan Huizing, PhD (2020-present)
George Karpati, MD, FRCP(C), FRS(C), OC; McGill University (2004-2009)
May Christine Malicdan, MD, PhD (2020-present)
Erin K O'Ferrall, MD, MSc, FRCPC; Montreal Neurological Institute (2009-2020)
Michael Sinnreich, MD, PhD; University Hospital Basel (2004-2020)
Revision History
- 9 April 2020 (bp) Comprehensive update posted live
- 7 March 2013 (me) Comprehensive update posted live
- 6 August 2009 (me) Comprehensive update posted live
- 24 May 2006 (me) Comprehensive update posted live
- 26 March 2004 (me) Review posted live
- 17 November 2003 (gk) Original submission
Note: Pursuant to 17 USC Section 105 of the United States Copyright Act, the GeneReview "GNE Myopathy" is in the public domain in the United States of America.
References
Literature Cited
- Jónsson H, Sulem P, Kehr B, Kristmundsdottir S, Zink F, Hjartarson E, Hardarson MT, Hjorleifsson KE, Eggertsson HP, Gudjonsson SA, Ward LD, Arnadottir GA, Helgason EA, Helgason H, Gylfason A, Jonasdottir A, Jonasdottir A, Rafnar T, Frigge M, Stacey SN, Th Magnusson O, Thorsteinsdottir U, Masson G, Kong A, Halldorsson BV, Helgason A, Gudbjartsson DF, Stefansson K. Parental influence on human germline de novo mutations in 1,548 trios from Iceland. Nature. 2017;549:519–22. [PubMed: 28959963]
- Arai A, Tanaka K, Ikeuchi T, Igarashi S, Kobayashi H, Asaka T, Date H, Saito M, Tanaka H, Kawasaki S, Uyama E, Mizusawa H, Fukuhara N, Tsuji S. A novel mutation in the GNE gene and a linkage disequilibrium in Japanese pedigrees. Ann Neurol. 2002;52:516–9. [PubMed: 12325084]
- Argov Z, Eisenberg I, Grabov-Nardini G, Sadeh M, Wirguin I, Soffer D, Mitrani-Rosenbaum S. Hereditary inclusion body myopathy: the Middle Eastern genetic cluster. Neurology. 2003;60:1519–23. [PubMed: 12743242]
- Argov Z, Yarom R. "Rimmed vacuole myopathy" sparing the quadriceps: a unique disorder in Iranian Jews. J Neurol Sci. 1984;64:33–43. [PubMed: 6737002]
- Bhattacharya S, Khadilkar SV, Nalini A, Ganapathy A, Mannan AU, Majumder PP, Bhattacharya A. Mutation spectrum of GNE myopathy in the Indian sub-continent. J Neuromuscul Dis. 2018;5:85–92. [PubMed: 29480215]
- Broccolini A, Gidaro T, De Cristofaro R, Morosetti R, Gliubizzi C, Ricci E, Tonali PA, Mirabella M. Hyposialylation of neprilysin possibly affects its expression and enzymatic activity in hereditary inclusion-body myopathy muscle. J Neurochem. 2008;105:971–81. [PubMed: 18182043]
- Broccolini A, Ricci E, Cassandrini D, Gliubizzi C, Bruno C, Tonoli E, Silvestri G, Pescatori M, Rodolico C, Sinicropi S, Servidei S, Zara F, Minetti C, Tonali PA, Mirabella M. Novel GNE mutations in Italian families with autosomal recessive hereditary inclusion-body myopathy. Hum Mutat. 2004;23:632. [PubMed: 15146476]
- Carrillo N, Malicdan MC, Huizing M. GNE myopathy: etiology, diagnosis, and therapeutic challenges. Neurotherapeutics. 2018;15:900–14. [PMC free article: PMC6277305] [PubMed: 30338442]
- Celeste FV, Vilboux T, Ciccone C, de Dios JK, Malicdan MC, Leoyklang P, McKew JC, Gahl WA, Carrillo-Carrasco N, Huizing M. Mutation update for GNE gene variants associated with GNE myopathy. Hum Mutat. 2014;35:915–26. [PMC free article: PMC4172345] [PubMed: 24796702]
- Cerino M, Gorokhova S, Béhin A, Urtizberea JA, Kergourlay V, Salvo E, Bernard R, Lévy N, Bartoli M, Krahn M. Novel pathogenic variants in a French cohort widen the mutational spectrum of GNE myopathy. J Neuromuscul Dis. 2015;2:131–6. [PMC free article: PMC5278624] [PubMed: 27858732]
- Chai Y, Bertorini TE, McGrew FA. Hereditary inclusion-body myopathy associated with cardiomyopathy: report of two siblings. Muscle Nerve. 2011;43:133–6. [PubMed: 21082694]
- Chamova T, Guergueltcheva V, Gospodinova M, Krause S, Cirak S, Kaprelyan A, Angelova L, Mihaylova V, Bichev S, Chandler D, Naydenov E, Grudkova M, Djukmedzhiev P, Voit T, Pogoryelova O, Lochmüller H, Goebel HH, Bahlo M, Kalaydjieva L, Tournev I. GNE myopathy in Roma patients homozygous for the p.I618T founder mutation. Neuromuscul Disord. 2015;25:713–8. [PubMed: 26231298]
- Chaouch A, Brennan KM, Hudson J, Longman C, McConville J, Morrison PJ, Farrugia ME, Petty R, Stewart W, Norwood F, Horvath R, Chinnery PF, Costigan D, Winer J, Polvikoski T, Healy E, Sarkozy A, Evangelista T, Pogoryelova O, Eagle M, Bushby K, Straub V, Lochmüller H. Two recurrent mutations are associated with GNE myopathy in the North of Britain. J Neurol Neurosurg Psychiatry. 2014;85:1359–65. [PMC free article: PMC6625961] [PubMed: 24695763]
- Chen Y, Xi J, Zhu W, Lin J, Luo S, Yue D, Cai S, Sun C, Zhao C, Mitsuhashi S, Nishino I, Xu M, Lu J. GNE myopathy in Chinese population: hotspot and novel mutations. J Hum Genet. 2019;64:11–16. [PubMed: 30390020]
- Cho A, Hayashi YK, Monma K, Oya Y, Noguchi S, Nonaka I, Nishino I. Mutation profile of the GNE gene in Japanese patients with distal myopathy with rimmed vacuoles (GNE myopathy). J Neurol Neurosurg Psychiatry. 2014;85:914–7. [PubMed: 24027297]
- Cohen M, Varki A. The sialome--far more than the sum of its parts. OMICS. 2010;14:455–64. [PubMed: 20726801]
- de Dios JK, Shrader JA, Joe GO, McClean JC, Williams K, Evers R, Malicdan MC, Ciccone C, Mankodi A, Huizing M, McKew JC, Bluemke DA, Gahl WA, Carrillo-Carrasco N. Atypical presentation of GNE myopathy with asymmetric hand weakness. Neuromuscul Disord. 2014;24:1063–7. [PMC free article: PMC4259851] [PubMed: 25182749]
- Del Bo R, Baron P, Prelle A, Serafini M, Moggio M, Fonzo AD, Castagni M, Bresolin N, Comi GP. Novel missense mutation and large deletion of GNE gene in autosomal-recessive inclusion-body myopathy. Muscle Nerve. 2003;28:113–7. [PubMed: 12811782]
- Effertz K, Hinderlich S, Reutter W. Selective loss of either the epimerase or kinase activity of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase due to site-directed mutagenesis based on sequence alignments. J Biol Chem. 1999;274:28771–8. [PubMed: 10497249]
- Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, Barash M, Shemesh M, Sadeh M, Grabov-Nardini G, Shmilevich I, Friedmann A, Karpati G, Bradley WG, Baumbach L, Lancet D, Asher EB, Beckmann JS, Argov Z, Mitrani-Rosenbaum S. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29:83–7. [PubMed: 11528398]
- Futterer J, Dalby A, Lowe GC, Johnson B, Simpson MA, Motwani J, Williams M, Watson SP, Morgan NV, et al. Mutation in GNE is associated with severe congenital thrombocytopenia. Blood. 2018;132:1855–8. [PMC free article: PMC6238157] [PubMed: 29941673]
- Gagiannis D, Orthmann A, Danssmann I, Schwarzkopf M, Weidemann W, Horstkorte R. Reduced sialylation status in UDP-N-acetylglucosamine-2-epimerase/N-acetylmannosamine kinase (GNE)-deficient mice. Glycoconj J. 2007;24:125–30. [PubMed: 17235685]
- Galeano B, Klootwijk R, Manoli I, Sun M, Ciccone C, Darvish D, Starost MF, Zerfas PM, Hoffmann VJ, Hoogstraten-Miller S, Krasnewich DM, Gahl WA, Huizing M. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest. 2007;117:1585–94. [PMC free article: PMC1878529] [PubMed: 17549255]
- Garland J, Stephen J, Class B, Gruber A, Ciccone C, Poliak A, Hayes CP, Singhal V, Slota C, Perreault J, Gavrilova R, Shrader JA, Chittiboina P, Joe G, Heiss J, Gahl WA, Huizing M, Carrillo N, Malicdan MCV. Identification of an Alu element-mediated deletion in the promoter region of GNE in siblings with GNE myopathy. Mol Genet Genomic Med. 2017;5:410–7. [PMC free article: PMC5511805] [PubMed: 28717665]
- Hackman P, Sarparanta J, Lehtinen S, Vihola A, Evilä A, Jonson PH, Luque H, Kere J, Screen M, Chinnery PF, Åhlberg G, Edström L, Udd B. Welander distal myopathy is caused by a mutation in the RNA-binding protein TIA1. Ann Neurol. 2013;73:500–9. [PubMed: 23401021]
- Harris-Love MO, Joe G, Davenport TE, Koziol D, Abbett Rose K, Shrader JA, Vasconcelos OM, McElroy B, Dalakas MC. Reliability of the adult myopathy assessment tool in individuals with myositis. Arthritis Care Res. 2015;67:563–70. [PMC free article: PMC4450351] [PubMed: 25201624]
- Huizing M, Malicdan MV, Krasnewich DM, Manoli I, Carrillo-Carrasco N. GNE myopathy. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, eds. The Metabolic and Molecular Bases of Inherited Disease. 8 ed. New York: McGraw-Hill; 2001.
- Huizing M, Carrillo-Carrasco N, Malicdan MC, Noguchi S, Gahl WA, Mitrani-Rosenbaum S, Argov Z, Nishino I. GNE myopathy: new name and new mutation nomenclature. Neuromuscul Disord. 2014;24:387–9. [PMC free article: PMC4015343] [PubMed: 24685570]
- Huizing M, Rakocevic G, Sparks SE, Mamali I, Shatunov A, Goldfarb L, Krasnewich D, Gahl WA, Dalakas MC. Hypoglycosylation of alpha-dystroglycan in patients with hereditary IBM due to GNE mutations. Mol Genet Metab. 2004;81:196–202. [PubMed: 14972325]
- Jebsen RH, Taylor N, Trieschmann RB, Trotter MJ, Howard LA. An objective and standardized test of hand function. Arch Phys Med Rehabil. 1969;50:311–9. [PubMed: 5788487]
- Keppler OT. UDP-GlcNAc 2-epimerase: a regulator of cell surface sialylation. Science. 1999;284:1372–6. [PubMed: 10334995]
- Kimonis VE, Fulchiero E, Vesa J, Watts G. VCP disease associated with myopathy, Paget disease of bone and frontotemporal dementia: review of a unique disorder. Biochim Biophys Acta. 2008;1782:744–8. [PubMed: 18845250]
- Khadilkar SV, Nallamilli BR, Bhutada A, Hegde M, Gandhi K, Faldu HD, Patil SB. A report on GNE myopathy: individuals of Rajasthan ancestry share the Roma gene. J Neurol Sci. 2017;375:239–40. [PubMed: 28320138]
- Kurochkina N, Yardeni T, Huizing M. Molecular modeling of the bifunctional enzyme UDP-GlcNAc 2-epimerase/ManNAc kinase and predictions of structural effects of mutations associated with HIBM and sialuria. Glycobiology. 2010;20:322–37. [PMC free article: PMC2815652] [PubMed: 19917666]
- Leoyklang P, Class B, Noguchi S, Gahl WA, Carrillo N, Nishino I, Huizing M, Malicdan MC. Quantification of lectin fluorescence in GNE myopathy muscle biopsies. Muscle Nerve. 2018;58:286–92. [PMC free article: PMC6105422] [PubMed: 29603301]
- Lochmüller H, Behin A, Caraco Y, Lau H, Mirabella M, Tournev I, Tarnopolsky M, Pogoryelova O, Woods C, Lai A, Shah J, Koutsoukos T, Skrinar A, Mansbach H, Kakkis E, Mozaffar T. A phase 3 randomized study evaluating sialic acid extended-release for GNE myopathy. Neurology. 2019;92:e2109–e2117. [PMC free article: PMC6512882] [PubMed: 31036580]
- Malicdan MC, Noguchi S, Hayashi YK, Nonaka I, Nishino I. Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat Med. 2009;15:690–5. [PubMed: 19448634]
- Mori-Yoshimura M, Oya Y, Hayashi YK, Noguchi S, Nishino I, Murata M. Respiratory dysfunction in patients severely affected by GNE myopathy (distal myopathy with rimmed vacuoles). Neuromuscul Disord. 2013;23:84–8. [PubMed: 23127962]
- Muelas N, Hackman P, Luque H, Garcés-Sánchez M, Azorín I, Suominen T, Sevilla T, Mayordomo F, Gómez L, Martí P, María Millán J, Udd B, Vílchez JJ. MYH7 gene tail mutation causing myopathic profiles beyond Laing distal myopathy. Neurology. 2010;75:732–41. [PubMed: 20733148]
- Niethamer TK, Yardeni T, Leoyklang P, Ciccone C, Astiz-Martinez A, Jacobs K, Dorward HM, Zerfas PM, Gahl WA, Huizing M. Oral monosaccharide therapies to reverse renal and muscle hyposialylation in a mouse model of GNE myopathy. Mol Genet Metab. 2012;107:748–55. [PMC free article: PMC3504164] [PubMed: 23122659]
- Nishino I, Noguchi S, Murayama K, Driss A, Sugie K, Oya Y, Nagata T, Chida K, Takahashi T, Takusa Y, Ohi T, Nishimiya J, Sunohara N, Ciafaloni E, Kawai M, Aoki M, Nonaka I. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology. 2002;59:1689–93. [PubMed: 12473753]
- Noguchi S, Keira Y, Murayama K, Ogawa M, Fujita M, Kawahara G, Oya Y, Imazawa M, Goto Y, Hayashi YK, Nonaka I, Nishino I. Reduction of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase activity and sialylation in distal myopathy with rimmed vacuoles. J Biol Chem. 2004;279:11402–7. [PubMed: 14707127]
- Nonaka I, Sunohara N, Ishiura S, Satoyoshi E. Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J Neurol Sci. 1981;51:141–55. [PubMed: 7252518]
- Olivé M, Goldfarb LG, Shatunov A, Fischer D, Ferrer I. Myotilinopathy: refining the clinical and myopathological phenotype. Brain. 2005;128:2315–26. [PubMed: 15947064]
- Park YE, Kim DS, Choi YC, Shin JH. Progression of GNE myopathy based on the patient-reported outcome. J Clin Neurol. 2019;15:275–84. [PMC free article: PMC6620453] [PubMed: 31286697]
- Penner J, Mantey LR, Elgavish S, Ghaderi D, Cirak S, Berger M, Krause S, Lucka L, Voit T, Mitrani-Rosenbaum S, Hinderlich S. Influence of UDP-GlcNAc 2-epimerase/ManNAc kinase mutant proteins on hereditary inclusion body myopathy. Biochemistry. 2006;45:2968–77. [PubMed: 16503651]
- Quintana M, Shrader J, Slota C, Joe G, McKew JC, Fitzgerald M, Gahl WA, Berry S, Carrillo N. Bayesian model of disease progression in GNE myopathy. Stat Med. 2019;38:1459–74. [PubMed: 30511500]
- Ricci E, Broccolini A, Gidaro T, Morosetti R, Gliubizzi C, Frusciante R, Di Lella GM, Tonali PA, Mirabella M. NCAM is hyposialylated in hereditary inclusion body myopathy due to GNE mutations. Neurology. 2006;66:755–8. [PubMed: 16534119]
- Sadeh M, Gadoth N, Hadar H, Ben-David E. Vacuolar myopathy sparing the quadriceps. Brain. 1993;116:217–32. [PubMed: 8453459]
- Sarparanta J, Jonson PH, Golzio C, Sandell S, Luque H, Screen M, McDonald K, Stajich JM, Mahjneh I, Vihola A, Raheem O, Penttilä S, Lehtinen S, Huovinen S, Palmio J, Tasca G, Ricci E, Hackman P, Hauser M, Katsanis N, Udd B. Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat Genet. 2012;44:450-5, S1-2. [PMC free article: PMC3315599] [PubMed: 22366786]
- Savarese M, Sarparanta J, Vihola A, Udd B, Hackman P. Increasing role of titin mutations in neuromuscular disorders. J Neuromuscul Dis. 2016;3:293–308. [PMC free article: PMC5123623] [PubMed: 27854229]
- Schwarzkopf M, Knobeloch KP, Rohde E, Hinderlich S, Wiechens N, Lucka L, Horak I, Reutter W, Horstkorte R. Sialylation is essential for early development in mice. Proc Natl Acad Sci U S A. 2002;99:5267–70. [PMC free article: PMC122758] [PubMed: 11929971]
- Selcen D, Engel AG. Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann Neurol. 2005;57:269–76. [PubMed: 15668942]
- Senderek J, Garvey SM, Krieger M, Guergueltcheva V, Urtizberea A, Roos A, Elbracht M, Stendel C, Tournev I, Mihailova V, Feit H, Tramonte J, Hedera P, Crooks K, Bergmann C, Rudnik-Schöneborn S, Zerres K, Lochmüller H, Seboun E, Weis J, Beckmann JS, Hauser MA, Jackson CE. Autosomal-dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am J Hum Genet. 2009;84:511–8. [PMC free article: PMC2667977] [PubMed: 19344878]
- Seppala R, Lehto VP, Gahl WA. Mutations in the human UDP-N-acetylglucosamine 2-epimerase gene define the disease sialuria and the allosteric site of the enzyme. Am J Hum Genet. 1999;64:1563–9. [PMC free article: PMC1377899] [PubMed: 10330343]
- Sparks SE, Ciccone C, Lalor M, Orvisky E, Klootwijk R, Savelkoul PJ, Dalakas MC, Krasnewich DM, Gahl WA, Huizing M. Use of a cell-free system to determine UDPN-acetylglucosamine 2-epimerase and N-acetylmannosamine kinase activities in human hereditary inclusion body myopathy. Glycobiology. 2005;15:1102–10. [PubMed: 15987957]
- Tajima Y, Uyama E, Go S, Sato C, Tao N, Kotani M, Hino H, Suzuki A, Sanai Y, Kitajima K, Sakuraba H. Distal myopathy with rimmed vacuoles: impaired O-glycan formation in muscular glycoproteins. Am J Pathol. 2005;166:1121–30. [PMC free article: PMC1602383] [PubMed: 15793292]
- Tasca G, Ricci E, Monforte M, Laschena F, Ottaviani P, Rodolico C, Barca E, Silvestri G, Iannaccone E, Mirabella M, Broccolini A. Muscle imaging findings in GNE myopathy. J Neurol. 2012;259:1358–65. [PubMed: 22231866]
- Udd B, Griggs R. Distal myopathies. Curr Opin Neurol. 2001;14:561–6. [PubMed: 11562566]
- Visser J, Mans E, de Visser M, van den Berg-Vos RM, Franssen H, de Jong JM, van den Berg LH, Wokke JH, de Haan RJ. Comparison of maximal voluntary isometric contraction and hand-held dynamometry in measuring muscle strength of patients with progressive lower motor neuron syndrome. Neuromuscul Disord. 2003;13:744–50. [PubMed: 14561498]
- Wopereis S, Lefeber DJ., Morava E, Wevers RA. Mechanisms in protein O-glycan biosynthesis and clinical and molecular aspects of protein O-glycan biosynthesis defects: a review. Clin Chem. 2006;52:574–600. [PubMed: 16497938]
- Xu X, Wang AQ, Latham LL, Celeste F, Ciccone C, Malicdan MC, Goldspiel B, Terse P, Cradock J, Yang N, Yorke S, McKew JC, Gahl WA, Huizing M, Carrillo N. Safety, pharmacokinetics and sialic acid production after oral administration of N-acetylmannosamine (ManNAc) to subjects with GNE myopathy. Mol Genet Metab. 2017;122:126–34. [PMC free article: PMC5949875] [PubMed: 28641925]
- Zhao J, Wang Z, Hong D, Lv H, Zhang W, Chen J, Yuan Y. Mutational spectrum and clinical features in 35 unrelated mainland Chinese patients with GNE myopathy. J Neurol Sci. 2015;354:21–26. [PubMed: 25986339]
- Zhu W, Mitsuhashi S, Yonekawa T, Noguchi S, Huei JC, Nalini A, Preethish-Kumar V, Yamamoto M, Murakata K, Mori-Yoshimura M, Kamada S, Yahikozawa H, et al. Missing genetic variations in GNE myopathy: rearrangement hotspots encompassing 5'UTR and founder allele. J Hum Genet. 2017;62:159–66. [PubMed: 27829678]
Publication Details
Author Information and Affiliations
Bethesda, Maryland
Bethesda, Maryland
Bethesda, Maryland
Publication History
Initial Posting: March 26, 2004; Last Update: April 9, 2020.
Copyright
GeneReviews® chapters are owned by the University of Washington. Permission is hereby granted to reproduce, distribute, and translate copies of content materials for noncommercial research purposes only, provided that (i) credit for source (http://www.genereviews.org/) and copyright (© 1993-2024 University of Washington) are included with each copy; (ii) a link to the original material is provided whenever the material is published elsewhere on the Web; and (iii) reproducers, distributors, and/or translators comply with the GeneReviews® Copyright Notice and Usage Disclaimer. No further modifications are allowed. For clarity, excerpts of GeneReviews chapters for use in lab reports and clinic notes are a permitted use.
For more information, see the GeneReviews® Copyright Notice and Usage Disclaimer.
For questions regarding permissions or whether a specified use is allowed, contact: ude.wu@tssamda.
Publisher
University of Washington, Seattle, Seattle (WA)
NLM Citation
Carrillo N, Malicdan MC, Huizing M. GNE Myopathy. 2004 Mar 26 [Updated 2020 Apr 9]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024.
