

NCBI Prokaryotic Genome Annotation Process
Go back to NCBI Prokaryotic Genome Annotation Pipeline
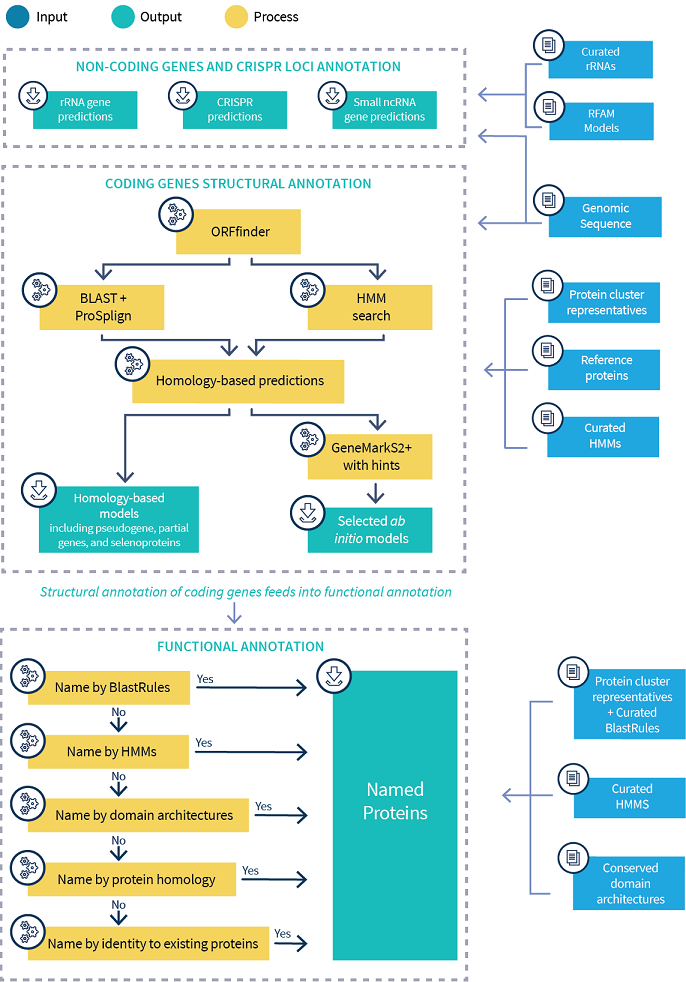
NCBI has developed a new approach to genome annotation that combines alignment-based methods with methods of predicting protein-coding and RNA genes and other functional elements directly from sequence. PGAP determines structural annotation by comparing open reading frames (ORFs) to libraries of protein hidden Markov models (HMMs), representative RefSeq proteins, and proteins from well characterized reference genomes. GeneMarkS-2+ then makes ab initio coding region predictions for genomic regions that lack HMM or protein evidence and selects start sites for ORFs whose evidence comes from HMMs.
The flowchart below describes the major components of the pipeline:
Structural annotation
Proteins
ORFs are predicted by ORFfinder in all six frames of the genome and searched against a library of HMMs (TIGRFAM, Pfam; PRK HMMs, and NCBIfams, a collection for high-value protein families, including proteins involved in antimicrobial resistance). Short ORFs without HMM hits that overlap with ORFs with hits are dropped. The remaining translated ORFs are searched against BlastRules, proteins from lineage-specific reference genomes and protein cluster representatives, using BLAST followed by ProSplign (ProSplign aligns proteins even in the presence of frameshifts). HMM hits and protein alignments are mapped from ORFs to the genome. The final set of predicted proteins is made based on the resulting aligning evidence, and the ab initio gene-finding program GeneMark-S2+, in regions that lack protein alignment evidence.
Note that the final annotation can contain programmed frameshifts/ribosomal slippage for some transposases and PrfB genes, etc. and provides a translated CDS feature for these genes. Selenoproteins are detected as well. Other frameshifts or internal stops are annotated as pseudo. PGAP also annotates partial genes when it cannot find start or stop for the evidence. Partial genes are translated when abutting sequence ends or gaps, or flagged as pseudo in the middle of the sequence.
Non-coding RNA
Structural RNAs/small ncRNAs
Structural RNAs (5S, 16S, and 23S rRNAs) and small non-coding RNAs are annotated by searching RFAM models against the query genome with the Infernal's cmsearch. 16S and 23S candidate features that span a mismatch of 100 bases or more are annotated as misc_feature rather than rRNA features.
tRNAs
To identify tRNA genes, the input genome sequence is split into ~200nt windows with overlap of ~100nt and passed through tRNAscan-SE. tRNAscan-SE identifies 99–100% of transfer RNA genes in DNA sequence while giving less than one false positive per 15 gigabases. It is currently one of the most powerful tRNA identification tools, and uses different, targeted parameter sets for Archaea and Bacteria. tRNA predictions below a tRNAscan-SE score of 20 are discarded.
Mobile/fast evolving genes
Phages
The annotation of phage related proteins is based on homology to a reference set of curated phage proteins. The phage reference data set comes from an independent effort of calculating and curating protein clusters from the complete bacteriophage genomes.
CRISPR
CRISPRs(Clustered Regularly Interspaced Short Palindromic Repeats) are a family of DNA direct repeats of 20 to 40 nucleotides separated by unique sequences of similar length and are commonly found in prokaryotic genomes. These defense systems are encoded by operons that have an extraordinarily diverse architecture and a high rate of evolution for both the cas genes and the unique spacer content. CRISPRs are identified by searching the CRISPR database with PILER-CR and CRISPR Recognition Tool (CRT). Piler-CR uses internal BLAST hits to identify repeat regions of suitable length. After that it does more sensitive search to find fragmented or degraded copies of the repeat family. CRT's search for CRISPRs is based on finding a series of short exact repeats of length k that are separated by a similar distance and then extending these exact k-mer matches to the actual repeat length.
Functional annotation
Predicted coding proteins are searched against Protein Family Models, a hierarchical collection of evidence composed of HMMs, BlastRules and domain architectures. Proteins are assigned the name and attributes (gene symbols, publications, and EC numbers if available) of the highest-precedence Protein Family Model that they hit (see more information about the naming process and the evidence collection). As a last resort, proteins that do not hit any evidence are named based on homology to protein cluster representatives. The names given to proteins follow the International Protein Nomenclature Guidelines, agreed upon by the European Bioinformatics Institute (EMBL-EBI), the National Center for Biotechnology Information (NCBI), the Protein Information Resource (PIR) and the Swiss Institute for Bioinformatics (SIB).
Annotation results
The annotation pipeline produces files ready for GenBank submission. All annotated sequences contain a summary of the annotation results for the entire assembly that includes the following:
- Annotation Provider: Organization that produced the annotation (typically NCBI)
- Annotation Date: Date/time the annotation was performed
- Annotation Pipeline: The pipeline used to create the annotation: NCBI Prokaryotic Genome Annotation Pipeline (PGAP)
- Annotation Method: PGAP uses a best-placed reference protein set and GeneMarkS-2+ for annotation, further details can be found here:Li W et al and Haft DH et al 2018
- Annotation Software revision: Version of the PGAP annotation software used. Release notes for each version can be found here: PGAP Release notes
- Features Annotated: Features that are annotated by PGAP: Genes, CDS, RNAs, etc.
- Genes (total): Total number of genes. Sum of Genes (coding), Genes (RNA) and Pseudo Genes (total)
- CDSs (total): Total number of coding sequences. Sum of CDSs (with protein) and CDSs (without protein)
- Genes (RNA): Total number of genes for the different classes of RNAs. Sum of genes for complete and partial rRNAs, tRNAs and ncRNAs
- rRNAs: Total number of the structural RNAs which make up the RNA component of the ribosome. For prokaryotes there are 3 types of ribosomal RNAs (rRNAs), 5S rRNA, 16S rRNA and 23S rRNA. The number of complete rRNAs for each type is followed by the type of rRNA in order. Example: 8, 7, 7 (5S, 16S, 23S)= 8 5S rRNA, 7 16S rRNA, 7 23S rRNA
- complete rRNAs: Total number of complete rRNAS for each type of rRNA
- partial rRNAs: Total number of partial rRNAs
- tRNAs: Total number of transfer RNAs
- ncRNAs: Total number of non-coding rRNAs
- Pseudo Genes (total): Total number of pseudo genes in the genome. These are genes that contain frameshifts or internal stops when translated. Partial genes that occur in the middle of a sequence are also flagged as pseudo.
- CDS (without protein): Total number of coding sequences which do not code for a protein
- Pseudo Genes (frameshifted): Total number of pseudo genes with frameshift(s) in the translated coding sequence
- Pseudo Genes (incomplete): Total number of pseudo genes which code for an incomplete protein
- Pseudo Genes (internal stop): Total number of pseudo genes which contain an internal stop codon in the translated coding sequence
- Pseudo Genes (multiple problems): Total number of pseudo genes which contain one or more problems in the above categories
- CRISPR Arrays: Total number of CRISPR elements annotated on the genome
Example of a summary report:
##Genome-Annotation-Data-START##
Annotation Provider :: NCBI
Annotation Date :: 09/07/2019 04:59:53
Annotation Pipeline :: NCBI Prokaryotic Genome Annotation Pipeline (PGAP)
Annotation Method :: Best-placed reference protein set; GeneMarkS-2+
Annotation Software revision :: 4.9
Features Annotated :: Gene; CDS; rRNA; tRNA; ncRNA
Genes (total) :: 5,913
CDSs (total) :: 5,784
Genes (coding) :: 5,522
CDSs (with protein) :: 5,522
Genes (RNA) :: 129
rRNAs :: 8, 7, 7 (5S, 16S, 23S)
complete rRNAs :: 8, 7, 7 (5S, 16S, 23S)
tRNAs :: 96
ncRNAs :: 11
Pseudo Genes (total) :: 262
CDSs (without protein) :: 262
Pseudo Genes (frameshifted) :: 118 of 262
Pseudo Genes (incomplete) :: 124 of 262
Pseudo Genes (internal stop) :: 66 of 262
Pseudo Genes (multiple problems):: 42 of 262
CRISPR Arrays) :: 2
##Genome-Annotation-Data-END##
References
Li W, O'Neill KR, Haft DH, DiCuccio M, Chetvernin V, Badretdin A, Coulouris G, Chitsaz F, Derbyshire MK, Durkin AS, Gonzales NR, Gwadz M, Lanczycki CJ, Song JS, Thanki N, Wang J, Yamashita RA, Yang M, Zheng C, Marchler-Bauer A, Thibaud-Nissen F. RefSeq: expanding the Prokaryotic Genome Annotation Pipeline reach with protein family model curation. Nucleic Acids Res. 2021 Jan 8;49(D1):D1020-D1028. doi: 10.1093/nar/gkaa1105. PMID: 33270901
Chan PP, Lowe TM. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol Biol. 2019;1962:1-14. doi: 10.1007/978-1-4939-9173-0_1. PMID: 31020551
Haft DH, DiCuccio M, Badretdin A, Brover V, Chetvernin V, O'Neill K, Li W, Chitsaz F, Derbyshire MK, Gonzales NR, Gwadz M, Lu F, Marchler GH, Song JS, Thanki N, Yamashita RA, Zheng C, Thibaud-Nissen F, Geer LY, Marchler-Bauer A, Pruitt KD. RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res. 2018 Jan 4;46(D1):D851-D860. doi: 10.1093/nar/gkx1068. PMID: 29112715
Lomsadze A, Gemayel K, Tang S, Borodovsky M. Modeling leaderless transcription and atypical genes results in more accurate gene prediction in prokaryotes. Genome Res. 2018 Jul;28(7):1079-1089. doi: 10.1101/gr.230615.117. Epub 2018 May 17. PMID: 29773659
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016 Jan 4;44(D1):D279-85. doi: 10.1093/nar/gkv1344. Epub 2015 Dec 15. PMID: 26673716
Nawrocki EP, Burge SW, Bateman A, Daub J, Eberhardt RY, Eddy SR, Floden EW, Gardner PP, Jones TA, Tate J, Finn RD. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015 Jan;43(Database issue):D130-7. doi: 10.1093/nar/gku1063. Epub 2014 Nov 11. PMID: 25392425
Haft DH, Selengut JD, Richter RA, Harkins D, Basu MK, Beck E. TIGRFAMs and Genome Properties in 2013. Nucleic Acids Res. 2013 Jan;41(Database issue):D387-95. doi: 10.1093/nar/gks1234. Epub 2012 Nov 28. PMID: 23197656
Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013 Nov 15;29(22):2933-5. doi: 10.1093/bioinformatics/btt509. Epub 2013 Sep 4. PMID: 24008419
Klimke W, Agarwala R, Badretdin A, Chetvernin S, Ciufo S, Fedorov B, Kiryutin B, O'Neill K, Resch W, Resenchuk S, Schafer S, Tolstoy I, Tatusova T. The National Center for Biotechnology Information's Protein Clusters Database. Nucleic Acids Res. 2009 Jan;37(Database issue):D216-23. doi: 10.1093/nar/gkn734. Epub 2008 Oct 21. PMID: 18940865
Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC, Hugenholtz P. CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics. 2007 Jun 18;8:209. doi: 10.1186/1471-2105-8-209. PMID: 17577412
Edgar RC. PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics. 2007 Jan 20;8:18. doi: 10.1186/1471-2105-8-18. PMID: 17239253