

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
SNP FAQ Archive [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2005-.
This publication is provided for historical reference only and the information may be out of date.

What is ascertainment bias, and how does it relate to the calculation of SNP frequencies?
Ascertainment bias is a term in population genetics that describes systematic deviations from an expected theoretical result attributable to the sampling processes used to find (ascertain) SNPs and measure (estimate) their population-specific allele frequencies.
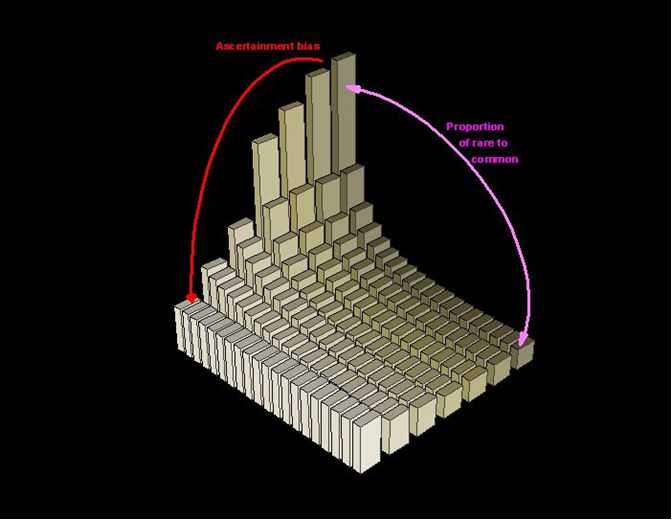
The distribution of SNP "derived" allele frequency ranges in nature from 1/2N (i.e., one mutant chromosome in the entire species gene pool), to 2N-1/2N (i.e., only one chromosome left in the species gene pool to represent the "ancestral" allelic state; all other chromosomes have the derived allele). This distribution, however, is imperfectly measured when finite sub-samples are drawn from the population. The smaller the finite sample used in our SNP detection (sampling) process, the more “imperfect” the fit between the distribution of derived allele frequencies and the “true” distribution in nature. Here is a graph that illustrates the bias:
Modified from Fig.2, Ref. (1).
If SNPs in dbSNP are ascertained in samples of a few chromosomes, then a fraction of those SNPs will be excessively common in the population relative to potentially larger samples of the same genomic sequence.
Experimental validation typically means observing the SNP in additional samples unrelated to the original set of chromosomes surveyed to define the SNP. Therefore, common SNPs will “validate” with a higher rate than SNPs with a really rare minor allele, because larger samples are needed to recapture (and hence confirm) the rare variation. Because all samples used to ascertain (discover) SNPs or estimate their allele frequencies in specific population samples are of finite size, there will be some kind of ascertainment bias in every batch of data submitted to dbSNP.
References
- 1.
Marth GT, Czabarka E, Murvai J, Sherry ST. (2004) The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics. Jan;166(1):351-372
- Ascertainment Bias - SNP FAQ ArchiveAscertainment Bias - SNP FAQ Archive
Your browsing activity is empty.
Activity recording is turned off.
See more...