
Copyright Notice
BLAST is a Registered Trademark of the National Library of Medicine
Bookshelf ID: NBK62051

An official website of the United States government

NCBI Bookshelf. A service of the National Library of Medicine, National Institutes of Health.
BLAST® Help [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2008-.
A fixed procedure embodied in a computer program.
The process or result of matching up the nucleotide or amino acid residues of two or more biological sequences to achieve maximal levels of identity and, in the case of amino acid sequences, conservation, for the purpose of assessing the degree of similarity and the possibility of homology.
Research, development, or application of computational tools and approaches for expanding the use of biological, medical, behavioral or health data, including those to acquire, store, organize, archive, analyze, or visualize such data.
The bit score, S', is derived from the raw alignment score, S, taking the statistical properties of the scoring system into account. Because bit scores are normalized with respect to the scoring system, they can be used to compare alignment scores from different searches.
Basic Local Alignment Search Tool (Altschul et al., 1990 & 1997) is a sequence comparison algorithm optimized for speed used to search sequence databases for optimal local alignments to a query. The initial search is done for a word of length "W" that scores at least "T" when compared to the query using a substitution matrix. Word hits are then extended in either direction in an attempt to generate an alignment with a score exceeding the threshold of "S". The "T" parameter dictates the speed and sensitivity of the search.

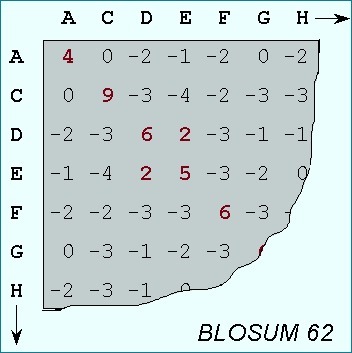
A Blocks Substitution Matrix is a substitution scoring matrix in which scores for each position are derived from observations of the frequencies of substitutions in blocks of local alignments in related proteins. Each matrix is tailored to a particular evolutionary distance. In the BLOSUM62 matrix, for example, the alignment from which scores were derived was created using sequences sharing no more than 62% identity. Sequences more identical than 62% are represented by a single sequence in the alignment so as to avoid over-weighting closely related family members. (Henikoff and Henikoff, 1992)
These are methods applied to protein BLAST searches that adjust the significance of alignment scores by taking into account the overall amino acid composition of the query and aligned database sequences. These methods provide more accurate statistics than those originally used in protein BLAST searches (Schäffer et al., 2001; Yu and Altschul, 2005). The conditional compositional score matrix adjustment method (Yu and Altschul, 2005) is used by default on the NCBI protein BLAST service.
A change at a specific position of an amino acid or, less commonly, DNA sequence that preserves the physico-chemical properties of the original residue or achieves a positive score in the governing scoring matrix.
A discrete portion of a protein assumed to fold independently of the rest of the protein and possessing its own function.
A program for filtering low complexity regions from nucleic acid sequences.
The Expectation value or Expect value represents the number of different alignments with scores equivalent to or better than S that is expected to occur in a database search by chance. The lower the E value, the more significant the score and the alignment.
The first widely used algorithm for database similarity searching. The program looks for optimal local alignments by scanning the sequence for small matches called "words". Initially, the scores of segments in which there are multiple word hits are calculated ("init1"). Later the scores of several segments may be summed to generate an "initn" score. An optimized alignment that includes gaps is shown in the output as "opt". The sensitivity and speed of the search are inversely related and controlled by the "k-tup" variable that specifies the size of a "word" (Pearson and Lipman, 1988).
Filtering, also known as masking, removes regions of (nucleic acid or amino acid) sequence having characteristics that may lead to spurious high scores. See SEG and DUST.
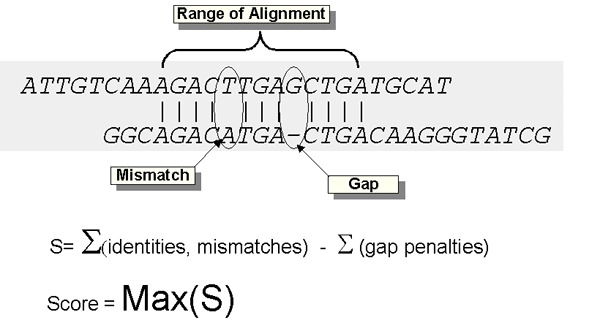
A space introduced into an alignment to compensate for insertions and deletions in one sequence relative to another. To prevent the accumulation of too many gaps in an alignment, introduction of a gap causes the deduction of a fixed amount (the gap score) from the alignment score. Extension of the gap to encompass additional nucleotides or amino acid is also penalized in the scoring of an alignment.
The alignment of two nucleic acid or protein sequences over their entire length.
H is the relative entropy of the target and background residue frequencies. (Karlin and Altschul, 1990). H can be thought of as a measure of the average information (in bits) available per position that distinguishes an alignment from chance. At high values of H short alignments can be distinguished by chance, whereas at lower H values a longer alignment may be necessary (Altschul, 1991).
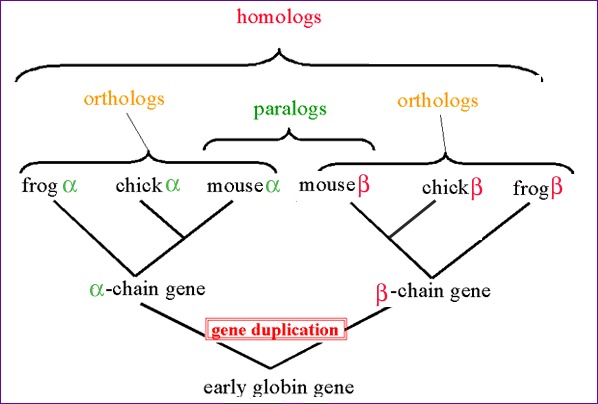
Similarity attributed to descent from a common ancestor. Homologous biological components (genes, proteins, structures) are called homologs. See also orthologs and paralogs.
A High-scoring Segment Pair (HSP) is a local alignment with no gaps that achieves one of the highest alignment scores in a given search.
The extent to which two (nucleotide or amino acid) sequences have the same residues at the same positions in an alignment, often expressed as a percentage.
A statistical parameter used in calculating BLAST scores that can be thought of as a natural scale for search space size. The value K is used in converting a raw score (S) to a bit score (S').
A statistical parameter used in calculating BLAST scores that can be thought of as a natural scale for scoring system. The value lambda is used in converting a raw score (S) to a bit score (S').
The alignment of a high-scoring region of two nucleic acid or protein sequences.
A region of biased composition in nucleic acid and protein sequences. These include homopolymeric runs, short-period repeats, and subtler over representation of one or a few residues. The SEG program is used to mask or filter low complexity regions in amino acid queries. The DUST program is used to mask or filter such regions in nucleic acid queries.
Also known as filtering. The removal of repeated or low complexity regions from a sequence in order to improve the sensitivity of sequence similarity searches performed with that sequence.
A short conserved region in a protein sequence. Motifs are frequently highly conserved parts of domains.
An alignment of three or more sequences with gaps inserted in the sequences such that residues with common structural positions and/or ancestral residues are aligned in the same column. Clustal W (Thompson, Higgins, and Gibson, 1994) is an example of a popular multiple sequence alignment program. The NCBI COBALT tool also produces multiple alignments of protein sequences (Papadopoulos and Agarwala, 2007).
An alignment of two sequences with the highest possible score.
Homologous biological components (genes, proteins, structures) in different species that arose from a single component present in the common ancestor of the species; orthologs may or may not have a similar function. Compare with paralogs.
The probability of a chance alignment occurring with a particular score or a better score in a database search. The p value is calculated by relating the observed alignment score, S, to the expected distribution of HSP scores from comparisons of random sequences of the same length and composition as the query to the database. The most highly significant P values will be those close to 0. P values and E values are different ways of representing the significance of the alignment.
Percent Accepted Mutation (PAM) is unit introduced by Margaret Dayhoff and colleagues to quantify the amount of evolutionary change in a protein sequence. 1.0 PAM unit is the amount of evolution that will change, on average, 1% of amino acids in a protein sequence. A PAM(x) substitution matrix is a look-up table in which scores for each amino acid substitution have been calculated based on the frequency of that substitution in closely related proteins that have experienced a certain amount (x) of evolutionary divergence.
Homologous biological components within a single species that arose by gene duplication. Compare with orthologs.
Position Hit Initiated BLAST (PHI-BLAST) is a variant of PSI-BLAST that can focus the alignment and construction of the PSSM around a motif, which must be present in the query sequence and is provided as input to the program. Only database sequences that contain the motif in context will be included in the results. See also PSSM.
A table that lists the frequencies of each amino acid in each position of protein sequence alignment. Frequencies are calculated from multiple alignments of sequences containing a domain of interest. See also PSSM.
Systematic analysis of protein expression of normal and diseased tissues that involves the separation, identification and characterization of all of the proteins in a sample.
Position-Specific Iterative BLAST (PSI-BLAST) is an iterative search using the protein BLAST algorithm. A profile is built after the initial search that is then used in subsequent searches. The process may be repeated, if desired, with new sequences found in each cycle used to refine the profile (Altschul et al., 1997).
A Position-Specific Scoring Matrix (PSSM) is a profile that gives the log-odds score for finding a particular matching amino acid in a target sequence.
The input sequence (or other type of search term) to which all of the entries in a database are to be compared.
The score of an alignment, S, calculated as the sum of substitution and gap scores. Substitution scores are given by a look-up table (see PAM, BLOSUM). Gap scores are typically calculated as the sum of G, the gap opening penalty and L, the gap extension penalty. For a gap of length n, the gap cost would be G+Ln. The choice of gap costs, G and L is empirical, but it is customary to choose a high value for G (10-15) and a low value for L (1-2).
The extent to which nucleotide or protein sequences are related. Similarity between two sequences can be expressed as percent sequence identity and/or percent positive substitutions.
A program for filtering low complexity regions in amino acid sequences (Wootton and Federhen, 1996). Residues that have been masked are represented as "X" in an alignment. SEG filtering is no longer the default in the NCBI blastp service because of the use of compositional adjustments to estimate BLAST statistics. See composition-based statistics.

The presence of a non-identical amino acid at a given position in an alignment. If the aligned residues have similar physico-chemical properties or have a positive score in the governing scoring matrix the substitution is said to be conservative.
A scoring matrix containing values proportional to the probability that amino acid i mutates into amino acid j for all pairs of amino acids. Such matrices are constructed by assembling a large and diverse sample of verified pairwise alignments of protein sequences. If the sample is large enough, the resulting matrices should reflect the true probabilities of mutations occurring through a period of evolution. The BLOSUM matrices are examples of substitution scoring matrices.
Also known as identity matrix. This is a scoring system in which only identical characters receive a positive score.
BLAST is a Registered Trademark of the National Library of Medicine
Your browsing activity is empty.
Activity recording is turned off.
See more...