X-Linked Sideroblastic Anemia and Ataxia – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY
Soumeya Bekri, Marc D'Hooghe, MD, and Pieter Vermeersch, MD, PhD.
Author Information and AffiliationsInitial Posting: March 1, 2006; Last Update: April 3, 2014.
Estimated reading time: 19 minutes
Summary
NOTE: THIS PUBLICATION HAS BEEN RETIRED. THIS ARCHIVAL VERSION IS FOR HISTORICAL REFERENCE ONLY, AND THE INFORMATION MAY BE OUT OF DATE.
Clinical characteristics.
X-linked sideroblastic anemia and ataxia (XLSA/A) is characterized by moderate anemia and early-onset spinocerebellar syndrome in males, manifest primarily as delayed walking, ataxia evident in early childhood, dysmetria, and dysdiadochokinesis. When present the intention tremor is mild and the dysarthria is mild to moderately severe. The ataxia has been described to be either non-progressive or slowly progressive. Upper motor neuron (UMN) signs in the legs, manifest by brisk deep tendon reflexes, unsustained ankle clonus, and equivocal or extensor plantar responses, are present in some males. Need for crutches or a wheelchair has been reported. Strabismus is seen in some males. Nystagmus and hypometric saccades may occur. Mild learning disability and depression are seen. The moderate hypochromic and microcytic anemia does not cause symptoms. Carrier (heterozygous) females have a normal neurologic examination and may show mild hematologic abnormalities.
Diagnosis/testing.
The diagnosis of XLSA/A is suspected in males with characteristic neurologic findings and the presence of moderate hypochromic and microcytic anemia, elevated whole blood total erythrocyte protoporphyrin (TEP) and zinc erythrocyte protoporphyrin (ZnEP), and ring sideroblasts on bone marrow examination. Pappenheimer bodies are seen in peripheral blood smear. The diagnosis is confirmed in a male by identification of a hemizygous pathogenic variant in ABCB7.
Females have a normal neurologic examination and may have a dimorphic blood smear with both hypochromic microcytic red blood cells and normal red blood cells; they may have ring sideroblasts on bone marrow examination.
Management.
Treatment of manifestations: Males with XLSA/A benefit from early physical therapy to facilitate acquisition of gross motor skills. Adaptive devices such as ankle fixation orthoses and walkers may be needed. Weighted eating utensils may help promote independent skills in childhood. Speech therapy may improve intelligibility problems from dysarthria. Difficulty with handwriting may be managed with computers for word processing.
Genetic counseling.
XLSA/A is inherited in an X-linked manner. Heterozygous females have a 50% chance of transmitting the pathogenic variant in each pregnancy. Males who inherit the pathogenic variant will be affected; females who inherit the pathogenic variant will be carriers and will usually not be affected. Males with XLSA/A will pass the pathogenic variant to all of their daughters and none of their sons. Carrier testing of at-risk female relatives and prenatal testing for a pregnancy at increased risk are possible if the ABCB7 pathogenic variant in the family is known.
Diagnosis
Males with X-linked sideroblastic anemia and ataxia (XLSA/A) exhibit the following signs:
Upper motor neuron (UMN) signs (i.e., brisk deep tendon reflexes, unsustained ankle clonus, and equivocal or extensor plantar responses) in the legs (present in some males)
Mild asymptomatic hypochromic, microcytic anemia
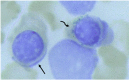
Ringed sideroblast Prussian blue staining of the bone marrow aspirate (x1000) showing a normal erythroid precursor (straight arrow) and a ringed sideroblast containing many iron granules around the nucleus (curved arrow).
Table 1.
Mean Corpuscular Volume (MCV fl) in XLSA/A
View in own window
| Group | Mean Corpuscular Volume (MCV fl) |
|---|
|
Normal male
| 89.1 ± 5.01 |
|
Normal female
| 87.6 ± 5.5 |
|
Affected male
| 58 to 68 |
|
Carrier female
| 83 to 90 |
Heterozygous females have a normal neurologic examination and may show mild hematologic abnormalities, including Pappenheimer bodies in peripheral blood smear [Hellier et al 2001] and ringed sideroblasts in bone marrow aspirate [Pagon et al 1985]. Some heterozygous females also have increased levels of whole blood TEP and ZnEP [Pagon et al 1985].
The diagnosis of XLSA/A is confirmed in a male by presence of a hemizygous pathogenic variant in ABCB7.
An alternative genetic testing strategy is use of a
multigene panel that includes
ABCB7 and other genes of interest (see
Differential Diagnosis). Note: The genes included and the methods used in multigene panels vary by laboratory and over time.
For an introduction to multigene panels click
here. More detailed information for clinicians ordering genetic tests can be found
here.
Table 2.
Molecular Genetic Testing Used in X-Linked Sideroblastic Anemia and Ataxia
View in own window
| Gene 1 | Method | Proportion of Male Probands with a Pathogenic Variant Detectable by Method |
|---|
|
ABCB7
| Sequence analysis 2, 3 | 4/4 families tested 4 |
| Deletion/duplication analysis 5 | Unknown, none reported 6 |
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
No deletions/duplications of ABCB7 have been reported to cause XLSA/A.
Clinical Characteristics
Clinical Description
To date, four unrelated families with X-linked sideroblastic anemia and ataxia (XLSA/A) have been reported [Allikmets et al 1999, Bekri et al 2000, Hellier et al 2001, Maguire et al 2001, D'Hooghe et al 2012].
Pagon et al [1985] reported two families with a non-progressive spinocerebellar syndrome and sideroblastic anemia, both segregating in an X-linked recessive mode of inheritance. Four males in two generations and a fifth male from an unrelated family were affected. Bekri et al [2000] reported two affected brothers with XLSA/A. Maguire et al [2001] reported another family with two affected brothers and two affected maternal uncles. D'Hooghe et al [2012] reported a boy with XLSA/A.
Ataxia/neurologic findings. Males have an early-onset spinocerebellar syndrome manifesting primarily as delayed walking, ataxia evident from early childhood, dysmetria, and dysdiadochokinesis. When present, intention tremor is mild and dysarthria is mild to moderately severe.
In some, but not all, affected members of one family, the ataxia appeared to improve with time, such that truncal titubation decreased and walking became progressively easier [Pagon et al 1985]. However, in older patients slow deterioration of walking can be seen in the fifth or sixth decade [Pagon et al 1985, Bekri et al 2000, Hellier et al 2001].
Need for crutches and/or a wheel chair have been reported.
Upper motor neuron (UMN) signs in the legs, manifest by brisk deep tendon reflexes, unsustained ankle clonus, and equivocal or extensor plantar responses are present in some males.
Strabismus is seen in some males. Extraocular movements are normal; however, nystagmus and hypometric saccades may occur.
Intellectual abilities are generally within the normal range. Mild learning disability and depression have been seen [Pagon & Bird, personal communication] and one person was reported to have "schizophrenia" [Hellier et al 2001].
Pes cavus, scoliosis, and muscle wasting are not present.
Impairment of visual acuity either from optic atrophy or retinal dystrophy is not seen.
In most cases brain MRI shows cerebellar atrophy/hypoplasia [Raskind et al 1991].
Anemia. The anemia is mild and does not cause symptoms.
Iron storage. Despite the finding of increased iron stores and ring sideroblasts on bone marrow examination, systemic iron overload has not been described. Serum iron studies including serum concentration of iron, total iron binding capacity (TIBC), per cent TIBC saturation, and serum concentration of ferritin were normal in the families reported by Pagon et al [1985] and Hellier et al [2001].
Heterozygotes. Carrier females have a normal neurologic examination.
Differential Diagnosis
Sideroblastic anemia. The sideroblastic anemias are a heterogeneous group of acquired and heritable anemias characterized by ringed sideroblasts (erythroid precursors - present in the bone marrow - that have pathologic iron overload in the mitochondria); the perinuclear location of mitochondria leads to the characteristic ringed appearance [Camaschella 2008, Bergmann et al 2010]. Iron inclusions, called Pappenheimer bodies, may also be observed in more mature erythrocytes [Sears & Udden 2004, Camaschella 2008] ().
The most common congenital sideroblastic anemia is X-linked sideroblastic anemia (XLSA) caused by mutation of ALAS2. XLSA is characterized by hepatic and systemic iron overload but not ataxia (as expression of ALAS2 is confined to erythroid tissues) [Napier et al 2005].
Other causes of congenital sideroblastic anemia include mutation of genes that encode proteins affecting mitochondrial metabolism [Bergmann et al 2010, Fujiwara & Harigae 2013] and/or affect iron-sulfur (Fe-S) cluster protein biosynthesis, assembly, or function. Fe-S proteins, which are essential for fundamental metabolic processes such as respiration and gene expression, are synthesized in the mitochondria and are either associated with mitochondrial apoprotein to form mitochondrial Fe/S proteins or exported to the cytosol (via the ABCB7 transporter) to assist in the formation of cytosolic and nuclear Fe-S proteins [Sheftel et al 2010]. These genes include the following:
SLC19A2 (encoding a high-affinity thiamine transporter)
ABCB7 (encoding mitochondrial ATP-binding cassette transporter), the cause of XLSA/A
GLRX5 (encoding monothiol glutaredoxin 5) [
Camaschella et al 2007], associated with a Fe-S cluster biosynthetic defect (the assembly of mitochondrial Fe/S proteins). The deregulation of mitochondrial iron metabolism results in sideroblastic anemia.
Mitochondrial DNA deletions, duplications and rearrangements (Pearson marrow-pancreas syndrome) [
Fleming 2002]
Another ataxia linked to Fe-S cluster protein is Friedreich ataxia (FRDA), caused by mutation of FXN, which encodes the mitochondrial protein frataxin whose main role is to supply iron in a bioavailable form for mitochondrial Fe-S cluster synthesis () [Ye & Rouault 2010b]. In persons with FRDA excess iron accumulation is observed in mitochondria of cardiac myocytes and neurons [Rouault & Tong 2008, Ye & Rouault 2010b]; however, persons with FRDA do not demonstrate significant anemia, suggesting either that frataxin is not essential for heme synthesis and erythropoiesis or that frataxin deficiency does not compromise erythropoietic tissues [Stemmler et al 2010, Ye & Rouault 2010a].
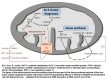
Fe-S cluster biogenesis, heme synthesis and hypothetic effects of defects in ABCB7 Adapted from D'Hooghe et al [2012] with permission
Of note, in murine cultured fibroblasts with decreased levels of frataxin, some Fe-S cluster proteins are deficient, iron accumulates in mitochondria, and oxidant sensitivity is observed as in the human disease [Calmels et al 2009]. The causal links among these effects are not well defined [Stemmler et al 2010].
Ataxia. The diagnostic approach to the numerous heritable ataxias can be quite challenging. Some hereditary ataxias typically present before age three years, including ataxia-telangiectasia, infantile-onset spinocerebellar ataxia, X-linked sideroblastic anemia with ataxia (XLSA/A), congenital disorders of glycosylation, and cerebellar malformations (e.g., Dandy-Walker malformation) [Bernard & Shevell 2008].
X-linked spinocerebellar ataxia has been reported, but is rare (see Hereditary Ataxia Overview). None has been associated with anemia. Mutation of ABCB7 should be considered in any unexplained X-linked spinocerebellar ataxia, even in the absence of clear hematologic changes.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease in an individual diagnosed with sideroblastic anemia and ataxia (XLSA/A), the following evaluations are recommended:
Treatment of Manifestations
There is no effective treatment for XLSA/A.
Males with ataxia benefit from early physical therapy to facilitate acquisition of gross motor skills. Adaptive devices such as ankle fixation orthoses and walkers may be needed.
Weighted eating utensils may help promote independent skills in childhood.
Speech therapy may improve intelligibility problems resulting from dysarthria.
Difficulty with handwriting may be managed with computers for word processing.
The anemia is usually mild and asymptomatic and does not require treatment; hepatic and systemic iron overload does not occur.
Surveillance
It is debated whether monitoring for possible iron overload is warranted in older individuals through routine screening of serum iron concentration, total iron binding capacity (TIBC), and serum ferritin concentration; iron overload is theoretically possible but has not been reported.
Therapies Under Investigation
One possible therapeutic approach to some of the disorders involving iron misdistribution is drug-mediated iron relocation. Deferiprone (DFP), an iron chelator used to treat iron overload, has iron-relocating abilities when used to treat disorders of regional iron accumulation. Because of their possible side effects, siderophores (small, high-affinity iron chelating compounds secreted by microorganisms) and other chelators must be administered with care; thus, the potential use of iron-redistributing agents in some iron-misdistribution diseases (such as XLSA/A) warrants rigorous investigation [Boddaert et al 2007, Camaschella 2008, Kakhlon et al 2010].
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Mode of Inheritance
X-linked sideroblastic anemia and ataxia (XLSA/A) is inherited in an X-linked manner.
Risk to Family Members
Parents of a proband
The father of an affected male will not have the disease nor will he be a
carrier of the pathogenic
ABCB7 allelic variant.
In a family with more than one affected individual, the mother of an affected male is an obligate
carrier.
If a woman has more than one affected son and the
pathogenic variant cannot be detected in her DNA, she has
germline mosaicism. To date germline mosaicism has not been reported in XLSA/A.
Sibs of a proband
The risk to sibs depends on the
carrier status of the mother.
If the mother of the
proband is a
carrier, the chance of transmitting the
pathogenic variant in each pregnancy is 50%. Male sibs who inherit the pathogenic
ABCB7 variant will be affected; female sibs who inherit the pathogenic variant will be carriers and will not be affected.
If the
pathogenic variant cannot be detected in the DNA of the mother of the only affected male in the family, the risk to sibs is low but greater than that of the general population because of the possibility of
germline mosaicism.
Offspring of a proband. Males with XLSA/A will pass the pathogenic variant to all of their daughters and none of their sons.
Other family members. The proband's maternal aunts may be at risk of being carriers and the aunt's offspring, depending on their gender, may be at risk of being carriers or of being affected.
Heterozygote (Carrier) Detection
Heterozygous females (carriers) are asymptomatic.
Carrier testing of at-risk female relatives is possible if the pathogenic variant has been identified in the family.
Prenatal Testing and Preimplantation Genetic Testing
Once the ABCB7 pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing, particularly if the testing is being considered for the purpose of pregnancy termination rather than early diagnosis. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
euro-ATAXIA (European Federation of Hereditary Ataxias)
Ataxia UK
Lincoln House, Kennington Park, 1-3 Brixton Road
London SW9 6DE
United Kingdom
Phone: +44 (0) 207 582 1444
Email: smillman@ataxia.org.uk
Medline Plus
National Ataxia Foundation
2600 Fernbrook Lane
Suite 119
Minneapolis MN 55447
Phone: 763-553-0020
Email: naf@ataxia.org
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
X-Linked Sideroblastic Anemia and Ataxia: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Pathogenesis
ABCB7 encodes a mitochondrial adenosine triphosphate (ATP)-binding cassette (ABC) transporter protein involved in iron homeostasis. The family of ABC transporters consists of a large group of ATP-dependent transmembrane proteins that specifically transport a wide variety of substrates across cell and organelle membranes [Holland 2011, Moitra & Dean 2011]. The human genome contains 49 ABC genes. Compared to bacteria, human mitochondria surprisingly harbor a very small number (≤4) of ABC transporters [Burke & Ardehali 2007, Zutz et al 2009]. Mitochondrial ABC transporters belong to the subfamily B and assemble as homodimers of half-transporters [Zutz et al 2009]. ABCB7 is found in the inner mitochondrial membrane.
The substrate transported by the ABCB7 transporter is not fully characterized. Initially, ABCB7 was thought to be involved in transport of heme from the mitochondria to the cytosol [Shimada et al 1998]. Recent studies in different species suggest relationships between heme biosynthetic pathways, iron-sulfur (Fe–S) cluster biogenesis, and mitochondrial iron homeostasis. The most common Fe–S clusters in eukaryotes are the [2Fe–2S] and [4Fe–4S] clusters. Fe–S clusters are ancient biologic prosthetic groups essential for numerous biologic processes, including mitochondrial respiratory chain activity and various other enzymatic and regulatory functions. Mitochondrial iron overload is a prominent feature of the human Fe–S cluster assembly disorders [Rouault & Tong 2008, Sheftel et al 2010]. It is possible that ABCB7 transports Fe-S clusters and/or an as-yet-unknown regulatory molecule from mitochondria to convey the signal that mitochondria have sufficient iron (). Then, if either Fe-S cluster synthesis or heme synthesis is disrupted, the cytosolic/nuclear compartment would perceive mitochondrial iron deficiency and could respond by significantly increasing mitochondrial iron stores.
See Additional information on pathogenesis (pdf).
Gene structure.
ABCB7 comprises 16 exons [Shimada et al 1998, Bekri et al 2000]. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. See Table 2 and Table 3. Only four unrelated families with XLSA/A have been reported, each with a distinct missense variant. It is worth noting that the identified pathogenic variants are missense variants with intermediate severity.
Bekri et al [2000] reported two affected brothers with XLSA/A and found a
hemizygous pathogenic
missense variant in
exon 10 (c.1300G>A; p.Glu434Lys) causing a substitution adjacent to the sixth putative transmembrane region of the ABCB7 protein; this variant was present in one
allele in their mother.
Maguire et al [2001] reported a family with two affected brothers and two affected maternal uncles. In the two brothers and in one uncle who was still alive,
Maguire et al [2001] found a
hemizygous pathogenic
missense variant in
exon 10 (c.1234G>C; p.Val412Leu) leading to a substitution in the last of six putative transmembrane regions of the ABCB7 protein. The mother was
heterozygote for this variant.
Table 3.
ABCB7 Pathogenic Variants Discussed in This GeneReview
View in own window
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|
| c.627A>T | p.Glu209Asp |
NM_004299.3
NP_004290.2
|
| c.1203T>G | p.Ile401Met |
| c.1234G>C | p.Val412Leu |
| c.1300G>A | p.Glu434Lys |
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
See Additional information on genetics of XLSA/A (pdf).
Normal gene product. The ATP-binding cassette, subfamily B, member 7 protein (ABCB7) belongs to the adenosine triphosphate-binding cassette transporter superfamily; its yeast ortholog, Atm1p, plays a central role in the maturation of cytosolic iron-sulfur (Fe-S) cluster-containing proteins [Bekri et al 2000]. ABCB7 contributes to the production of heme during the differentiation of erythroid cells [Taketani et al 2003]. It is also thought to transport a component required for the maturation of cytosolic Fe-S clusters from the mitochondrion to the cytosol [Napier et al 2005]. Thus, the mitochondrion appears to be important in both heme synthesis and in the biogenesis of Fe-S clusters.
ABCB7 is highly expressed in bone marrow as well as in the cerebellum, which may explain why males with ABCB7 deficiency have ataxia [Allikmets et al 1999, Ye & Rouault 2010a, Ye & Rouault 2010b]. The ataxia observed in XLSA/A may be related to the damage mediated by the iron loading in the mitochondrion and/or disruption to mitochondrial iron homeostasis in neural cells [Napier et al 2005].
Abnormal gene product. Complementation studies in yeast suggest that the human mutated ATP-binding cassette, subfamily B, member 7 proteins (ABCB7) are caused by mild, partial loss-of-function alleles [Allikmets et al 1999, Bekri et al 2000] that result in diminished cytosolic Fe-S cluster protein.
Pondarré et al [2006] created a conditional knockout allele of the murine ortholog Abcb7 and formally demonstrated that XLSA/A is caused by partial-loss-of-function variants that directly or indirectly inhibit heme biosynthesis [Pondarré et al 2007]. Indeed, mutation leading to a significant loss of protein function must be lethal as illustrated by a knockout mouse model [Pondarré et al 2006].
References
Literature Cited
Allikmets R, Raskind WH, Hutchinson A, Schueck ND, Dean M, Koeller DM. Mutation of a putative mitochondrial iron transporter gene (ABC7) in X-linked sideroblastic anemia and ataxia (XLSA/A).
Hum Mol Genet. 1999 May;8:743–9. [
PubMed: 10196363]
Bekri S, Kispal G, Lange H, Fitzsimons E, Tolmie J, Lill R, Bishop DF. Human ABC7 transporter: gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation.
Blood. 2000;96:3256–64. [
PubMed: 11050011]
Bergmann AK, Campagna DR, McLoughlin EM, Agarwal S, Fleming MD, Bottomley SS, Neufeld EJ. Systematic molecular genetic analysis of congenital sideroblastic anemia: evidence for genetic heterogeneity and identification of novel mutations.
Pediatr Blood Cancer. 2010 Feb;54:273–8. [
PMC free article: PMC2843911] [
PubMed: 19731322]
Bernard G, Shevell M. The wobbly child: an approach to inherited ataxias.
Semin Pediatr Neurol. 2008;15:194–208. [
PubMed: 19073328]
Boddaert N, Le Quan Sang KH, Rötig A, Leroy-Willig A, Gallet S, Brunelle F, Sidi D, Thalabard JC, Munnich A, Cabantchik ZI. Selective iron chelation in Friedreich ataxia: biologic and clinical implications.
Blood. 2007 Jul 1;110:401–8. [
PubMed: 17379741]
Boultwood J, Pellagatti A, Nikpour M, Pushkaran B, Fidler C, Cattan H, Littlewood TJ, Malcovati L, Della Porta MG, Jädersten M, Killick S, Giagounidis A, Bowen D, Hellström-Lindberg E, Cazzola M, Wainscoat JS. The role of the iron transporter ABCB7 in refractory anemia with ring sideroblasts.
PLoS ONE. 2008;3:e1970. [
PMC free article: PMC2276313] [
PubMed: 18398482]
Burke MA, Ardehali H. Mitochondrial ATP-binding cassette proteins.
Transl Res. 2007;150:73–80. [
PubMed: 17656326]
Bykhovskaya Y, Casas K, Mengesha E, Inbal A, Fischel-Ghodsian N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA).
Am J Hum Genet. 2004;74:1303–8. [
PMC free article: PMC1182096] [
PubMed: 15108122]
Calmels N, Schmucker S, Wattenhofer-Donzé M, Martelli A, Vaucamps N, Reutenauer L, Messaddeq N, Bouton C, Koenig M, Puccio H. The first cellular models based on frataxin missense mutations that reproduce spontaneously the defects associated with Friedreich ataxia.
PLoS One. 2009 Jul 24;4:e6379. [
PMC free article: PMC2710521] [
PubMed: 19629184]
Camaschella C. Recent advances in the understanding of inherited sideroblastic anaemia.
Br J Haematol. 2008;143:27–38. [
PubMed: 18637800]
Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, Levi S, Iolascon A. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload.
Blood. 2007;110:1353–8. [
PubMed: 17485548]
D'Hooghe M, Selleslag D, Mortier G, Van Coster R, Vermeersch P, Billiet J, Bekri S. X-linked sideroblastic anemia and ataxia: a new family with identification of a fourth ABCB7 gene mutation.
Eur J Paediatr Neurol. 2012 Nov;16:730–5. [
PubMed: 22398176]
Fleming MD. The genetics of inherited sideroblastic anemias.
Semin Hematol. 2002;39:270–81. [
PubMed: 12382202]
Fujiwara T, Harigae H. Pathophysiology and genetic mutations in congenital sideroblastic anemia.
Pediatr Int. 2013;55:675–9. [
PubMed: 24003969]
Guernsey DL, Jiang H, Campagna DR, Evans SC, Ferguson M, Kellogg MD, Lachance M, Matsuoka M, Nightingale M, Rideout A, Saint-Amant L, Schmidt PJ, Orr A, Bottomley SS, Fleming MD, Ludman M, Dyack S, Fernandez CV, Samuels ME. Mutations in mitochondrial carrier family gene SLC25A38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia.
Nat Genet. 2009 Jun;41:651–3. [
PubMed: 19412178]
Hart D, Piomelli S. Simultaneous quantitation of zinc protoporphyrin and free protoporphyrin in erythrocytes by acetone extraction.
Clin Chem. 1981;27:220–2. [
PubMed: 7460270]
Hellier KD, Hatchwell E, Duncombe AS, Kew J, Hammans SR. X-linked sideroblastic anaemia with ataxia: another mitochondrial disease?
J Neurol Neurosurg Psychiatry. 2001;70:65–9. [
PMC free article: PMC1763461] [
PubMed: 11118249]
Holland IB. ABC transporters, mechanisms and biology: an overview.
Essays Biochem. 2011;50:1–17. [
PubMed: 21967049]
Kakhlon O, Breuer W, Munnich A, Cabantchik ZI. Iron redistribution as a therapeutic strategy for treating diseases of localized iron accumulation.
Can J Physiol Pharmacol. 2010;88:187–96. [
PubMed: 20393584]
Maguire A, Hellier K, Hammans S, May A. X-linked cerebellar ataxia and sideroblastic anaemia associated with a missense mutation in the ABC7 gene predicting V411L.
Br J Haematol. 2001;115:910–7. [
PubMed: 11843825]
Moitra K, Dean M. Evolution of ABC transporters by gene duplication and their role in human disease.
Biol Chem. 2011;392:29–37. [
PubMed: 21194360]
Napier I, Ponka P, Richardson DR. Iron trafficking in the mitochondrion: novel pathways revealed by disease.
Blood. 2005;105:1867–74. [
PubMed: 15528311]
Nikpour M, Scharenberg C, Liu A, Conte S, Karimi M, Mortera-Blanco T, Giai V, Fernandez-Mercado M, Papaemmanuil E, Högstrand K, Jansson M, Vedin I, Stephen Wainscoat J, Campbell P, Cazzola M, Boultwood J, Grandien A, Hellström-Lindberg E. The transporter ABCB7 is a mediator of the phenotype of acquired refractory anemia with ring sideroblasts.
Leukemia. 2013;27:889–96. [
PMC free article: PMC3794445] [
PubMed: 23070040]
Piomelli S, Lamola AA, Poh-Fitzpatrick MF, Seaman C, Harber LC. Erythropoietic protoporphyria and lead intoxication: the molecular basis for difference in cutaneous photosensitivity. I. Different rates of disappearance of protoporphyrin from the erythrocytes, both in vivo and in vitro.
J Clin Invest. 1975;56:1519–27. [
PMC free article: PMC333130] [
PubMed: 1202082]
Pondarré C, Antiochos BB, Campagna DR, Clarke SL, Greer EL, Deck KM, McDonald A, Han AP, Medlock A, Kutok JL, Anderson SA, Eisenstein RS, Fleming MD. The mitochondrial ATP-binding cassette transporter Abcb7 is essential in mice and participates in cytosolic iron-sulphur cluster biogenesis.
Hum Mol Genet. 2006;15:953–64. [
PubMed: 16467350]
Pondarré C, Campagna DR, Antiochos B, Sikorski L, Mulhern H, Fleming MD. Abcb7, the gene responsible for X-linked sideroblastic anemia with ataxia, is essential for hematopoiesis.
Blood. 2007;109:3567–9. [
PMC free article: PMC1852240] [
PubMed: 17192398]
Raskind WH, Wijsman E, Pagon RA, Cox TC, Bawden MJ, May BK, Bird TD. X-linked sideroblastic anemia and ataxia: linkage to phosphoglycerate kinase at Xq13.
Am J Hum Genet. 1991;48:335–41. [
PMC free article: PMC1683027] [
PubMed: 1671320]
Riley LG, Cooper S, Hickey P, Rudinger-Thirion J, McKenzie M, Compton A, Lim SC, Thorburn D, Ryan MT, Giegé R, Bahlo M, Christodoulou J. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia--MLASA syndrome.
Am J Hum Genet. 2010 Jul 9;87:52–9. [
PMC free article: PMC2896778] [
PubMed: 20598274]
Sears DA, Udden MM. Pappenheimer bodies: a brief historical review.
Am J Hematol. 2004;75:249–50. [
PubMed: 15054821]
Sheftel A, Stehling O, Lill R. Iron-sulfur proteins in health and disease.
Trends Endocrinol Metab. 2010;21:302–14. [
PubMed: 20060739]
Shimada Y, Okuno S, Kawai A, Shinomiya H, Saito A, Suzuki M, Omori Y, Nishino N, Kanemoto N, Fujiwara T, Horie M, Takahashi E. Cloning and chromosomal mapping of a novel ABC transporter gene (hABC7), a candidate for X-linked sideroblastic anemia with spinocerebellar ataxia.
J Hum Genet. 1998;43:115–22. [
PubMed: 9621516]
Taketani S, Kakimoto K, Ueta H, Masaki R, Furukawa T. Involvement of ABC7 in the biosynthesis of heme in erythroid cells: interaction of ABC7 with ferrochelatase.
Blood. 2003;101:3274–80. [
PubMed: 12480705]
Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawahata R, Yamaguchi T, Otsu M, Obara N, Sakata-Yanagimoto M, Ishiyama K, Mori H, Nolte F, Hofmann WK, Miyawaki S, Sugano S, Haferlach C, Koeffler HP, Shih LY, Haferlach T, Chiba S, Nakauchi H, Miyano S, Ogawa S. Frequent pathway mutations of splicing machinery in myelodysplasia.
Nature. 2011;478:64–9. [
PubMed: 21909114]
Zutz A, Gompf S, Schagger H, Tampe R. Mitochondrial ABC proteins in health and disease.
Biochim Biophys Acta. 2009;1787:681–90. [
PubMed: 19248758]
Chapter Notes
Author History
Soumeya Bekri (2014-present)
Marc D'Hooghe, MD (2014-present)
Thomas D Bird, MD; University of Washington, Seattle (1998-2014)
Roberta A Pagon, MD; University of Washington, Seattle (1998-2014)
Pieter Vermeersch (2014-present)
Revision History
20 August 2020 (ma) Chapter retired: extremely rare disorder
3 April 2014 (me) Comprehensive update posted live
7 April 2009 (me) Comprehensive update posted live
1 May 2008 (cd) Revision: sequencing of exons 5-16 and the
intron/
exon junctions available clinically
24 March 2008 (cd) Revision: clinical testing not available
1 March 2006 (me) Review posted live
12 November 1998 (bp) Original submission