Clinical Description
Spinocerebellar ataxia type 7 (SCA7) comprises a phenotypic spectrum ranging from adolescent- or adult-onset progressive cerebellar ataxia and cone-rod retinal dystrophy with progressive central visual loss to infantile or early-childhood onset with multiorgan failure, an accelerated course, and early death [Giunti et al 1999].
One important aspect of SCA7 clinical manifestations is their extreme variability with respect to age of onset and rate of progression. Affected individuals may present in infancy, childhood, adolescence, young adulthood, middle age, or old age.
When onset is at or before adolescence, initial manifestations are typically impaired vision, ultimately progressing to blindness from retinal degeneration. Individuals with manifestations in their teens may be blind within a decade or less.
In adults, the progressive cerebellar ataxia (i.e., dysmetria, dysdiadochokinesia, and poor coordination) usually precedes the onset of visual manifestations. The age of onset inversely correlates with rate of progression and extent of symptomatology, as onset in or after the fifth decade of life gives a predominant cerebellar ataxia without progression to significant visual impairment, whereas onset prior to middle age often features progression to vision loss.
Progression to severe disability resulting in death varies based on age of onset, ranging from months in infants to fewer than ten years in older children to two to three decades in adolescents and adults. While the rate of progression varies, the eventual result for almost all affected individuals is severe dysarthria, dysphagia, and a bedridden state with loss of motor control.
To date, more than 1,000 individuals with SCA7 have been identified worldwide. Frequency of select features in adolescent- or adult-onset disease are summarized in Table 2.
Adolescent- or Adult-Onset SCA7
Table 2.
Select Features of Adolescent- or Adult-Onset SCA7
View in own window
| Feature | % of Persons w/Feature | Comment |
|---|
|
Cerebellar ataxia
| 100% | Unsteady gait; finger-to-nose dysmetria |
|
Dysarthria
| 100% | Garbled or slurred speech |
|
Dysphagia
| 40% | Difficulty swallowing |
Oculomotor
abnormalities
| 80% |
Slowed ocular saccades Ophthalmoplegia
|
Motor neuron
degeneration
| 100% |
Upper motor neuron involvement (hyperreflexia, spasticity); may resemble hereditary spastic paraplegia. Lower motor neuron involvement (fasciculations, weakness w/muscle wasting, areflexia, distal sensory loss)
|
|
Sensory loss
| 40% | ↓ sensation to light touch, pinprick, &/or joint position |
Restless leg
syndrome
| 35% | Discomfort in legs resulting in uncontrollable urge to move one’s legs, typically worse in evening or nighttime |
|
Cognitive decline
| 20% | Impaired executive function |
Behavior disorder/
Psychosis
| 10% |
Altered mentation Impaired reality testing
|
Cone-rod
dystrophy
| 70% |
|
Neurologic findings. In adult-onset disease (age >30 years), cerebellar ataxia (manifesting as difficulty with walking, manual dexterity, and speech) is the most common clinical feature and is often the first reported manifestation (see Genotype-Phenotype Correlations). Affected individuals often then develop more extensive neurologic deficits, dysarthria, dysphagia, hypoacusis (hearing loss), and eye movement abnormalities (slow ocular saccades, staring). Slowing of ocular saccades may progress to frank ophthalmoplegia.
Involvement of the corticospinal tracts, resulting in brisk tendon reflexes and spasticity, may become evident as the disease progresses.
Cognitive decline and psychosis have been reported [Benton et al 1998]. Neuropsychiatric testing of some individuals has revealed selective deficits in social cognition [Sokolovsky et al 2010].
Retinal degeneration. The retinal degeneration is a progressive cone-rod dystrophy that may result in total blindness [To et al 1993, Aleman et al 2002, Ahn et al 2005, Hugosson et al 2009].
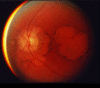
In adolescent- or young adult-onset disease (age <30 years), profound visual loss can be accompanied by minimal ophthalmoscopic findings and minimal ataxia [Thurtell et al 2009] (see Genotype-Phenotype Correlations). The onset of cone-rod dystrophy is often characterized by hemeralopia (inability to see clearly in bright light), photophobia (extreme sensitivity to light), decreased central visual acuity, and abnormalities in the tritan (blue-yellow) axis on detailed color vision testing [Miller et al 2009]. As cone function decreases over time, central visual acuity decreases to 20/200 (legally blind) and central scotomas develop; more prominent macular changes appear (see ), and all color discrimination is lost. Eventually all vision is lost.
Funduscopic photo shows extreme macular degeneration of late-stage SCA7.
Early signs of cone-rod dystrophy are subtle granular changes in the macula. Electroretinogram is consistently abnormal early in the disease course, showing a decrease in the photopic (cone) response initially, followed by a decrease in the scotopic (rod) response [Miller et al 2009].
In classic adult-onset disease (age >40 years), vision loss from retinal degeneration typically follows the onset of ataxia (sometimes many years to decades later) and gradually declines, seldom progressing to total blindness [Miller et al 2009].
Infantile- or Early Childhood-Onset SCA7
In infancy or early childhood disease, progression is always more rapid and aggressive than in adults. In infants, the clinical diagnosis may be elusive because ataxia and visual loss are not obvious; failure to thrive and loss of motor milestones may be the earliest findings. Other findings include progressive hypotonia, poor feeding, dysphagia, and congestive heart failure [Babovic-Vuksanovic et al 1998, Benton et al 1998]. Indeed, with rapid multisystem failure (including cerebellar and brain stem degeneration and other organ systems including lungs, heart, and kidneys), retinal degeneration and related vision loss may not be evident.
Affected infants usually die within months of initial presentation and never survive into early childhood [Ansorge et al 2004], a distinctly different clinical course from adult-onset SCA7, in which other organ system involvement does not occur (see Genotype-Phenotype Correlations).
Pathology. Neuronal loss, loss of myelinated fibers, and gliosis are observed in the cerebellum (especially Purkinje cells); the inferior olivary, dentate, and pontine nuclei; and to a lesser extent in the cerebral cortex, basal ganglia, thalamus, and midbrain [Rüb et al 2008, Seidel et al 2012].
Genotype-Phenotype Correlations
A correlation between CAG repeat sizes and disease severity exists: the longer the CAG repeat, the earlier the age of onset and the more severe and rapidly progressive the disease.
A correlation between CAG repeat size and initial clinical manifestation exists [Johansson et al 1998]:
CAG repeat sizes greater than 59 are typically associated with adolescent or young-adult onset (age <30 years) and visual impairment as the initial manifestation.
CAG repeat sizes smaller than 59 are often associated with adult onset (age >30 years) and cerebellar findings as the initial manifestation.
Despite observations correlating CAG repeat length with age of onset, disease severity, and course, CAG repeat size cannot provide sufficient predictive value for clinical prognosis within the classic adult-onset CAG repeat size range of 38 to 50 repeats [Andrew et al 1997].
Reports of pathogenic (age-related reduced-penetrance) repeats include the following:
Anticipation
In families with a pathogenic (full-penetrance) CAG repeat expansion, the repeat size tends to expand with transmission to successive generations, with more marked expansions seen in affected offspring of affected males [Gouw et al 1998]. This explains, at the genetic level, the marked anticipation seen in families with SCA7, now regarded as the most unstable of all CAG repeat disorders.
Anticipation in a family may be so dramatic that a child may be diagnosed with what is thought to be an unrelated neurodegenerative disease years before a parent or grandparent with pathogenic CAG repeat expansion becomes symptomatic [van de Warrenburg et al 2001, Ansorge et al 2004].
Repeat contraction has not been reported.
Prevalence
The prevalence is fewer than 1:300,000. In several studies, SCA7 represented 2% of all SCAs [Filla et al 2000, Storey et al 2000].
SCA7 occurs predominantly in two racial population groups: northern Europeans and Africans. Indeed, SCA7 is the only repeat expansion disease, with the exception of Huntington disease-like 2 (HDL2), with a large number of affected individuals of African racial ancestry. For this reason, a substantial fraction of individuals with SCA7 in the United States are of African racial ancestry. Worldwide, SCA7 is seen in North America, Europe, Eurasia, Australia, South Africa, and South America.
As a result of a founder effect in Mexico dating back to the colonial era, a very large concentration of individuals with SCA7 have been ascertained in the state of Veracruz in Mexico, with well over 150 documented affected individuals.