Clinical Description
Osteoglophonic dysplasia (OGD) is a skeletal dysplasia characterized by multisuture craniosynostosis (including premature fusion of the coronal, sagittal, lambdoid, and metopic sutures), distinctive craniofacial features, unerupted teeth, profound short stature, and multiple cystic bone lesions consistent with non-ossifying fibromas. To date, 24 individuals with OGD from 19 families have been reported and/or identified; of these, 14 individuals have had molecular genetic testing with a pathogenic variant identified in FGFR1 [Marzin et al 2020, Zou et al 2022]. The remaining ten individuals did not have molecular testing. The following description of the phenotypic features associated with this condition is based on these reports.
Table 2.
Osteoglophonic Dysplasia: Frequency of Select Features
View in own window
| Feature | Proportion of Persons w/Feature | Comment |
|---|
|
Non-ossifying fibromas
| 21/24 | Proximal & distal femur, distal tibia & fibula, iliac bones, proximal humerus, & distal radius & ulna |
|
Unerupted teeth
| 20/24 | |
|
Short stature
| 18/24 | |
|
Multisuture craniosynostosis
| 17/24 | Premature fusion of the coronal, sagittal, lambdoid, & metopic sutures |
|
Platyspondyly
| 10/24 | |
|
Hypophosphatemia
| 4/24 | Mediated by FGF23, a phosphaturic factor |
|
Giant cell granuloma of the jaw
| 3/24 | |
|
Increased body temperature
| 2/24 | Associated with excessive sweating & increased sensitivity to heat |
|
Overlapping toes
| 2/24 | 3rd overlapping or underriding 2nd & 4th |
|
Pyloric stenosis
| 2/24 | |
|
Inguinal hernia
| 2/24 | |
|
Choanal atresia/narrowing
| 2/24 | |
FGF23 = fibroblast growth factor 23
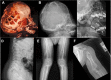
Skeletal. Multiple cystic lesions consistent with non-ossifying fibromas are seen on radiologic images of the proximal and distal femur, distal tibia and fibula, iliac bones, proximal humerus, and distal radius and ulna (, ). The cystic lesions appear early in life and gradually increase in size and number during childhood; later, they gradually ossify, regress, or disappear after skeletal maturity. The tubular bones appear broad and short with marked dysplastic changes at the epiphyseal ossification centers. Spinal imaging shows platyspondyly with anterior beaking and posterior scalloping of the lower thoracic and lumbar vertebral bodies () [Kelley et al 1983, Azouz & Kozlowski 1997, Sargar et al 2017, Marzin et al 2020]. Other skeletal features include rhizomelic limb shortening, short, broad hands and feet, genu varum, pseudoarthrosis, pathologic fractures, and overlapping toes, where the 3rd toe is overlapping or underriding the 2nd and 4th toes [A Othman, H Babcock, & C Ferreira, personal observations].
Growth. Individuals with OGD show impaired postnatal growth. Affected infants are below the 3rd centile for length but profound short stature becomes more evident with age. Rhizomelic limb shortening becomes increasingly apparent in childhood. Milder short stature has been reported in some individuals with normal to low normal height; adult height ranges between 97 and 154 cm [Beighton 1989, Marzin et al 2020].
Poor weight gain can be attributed to feeding difficulties, choanal atresia, or nasal obstruction with airway and breathing problems, which can occur during infancy as a result of craniofacial abnormalities and may rarely lead to death [Santos et al 1988].
Craniofacial. Multisuture craniosynostosis is present in most individuals. Head shape depends on the sutures involved and the timing of premature fusion, ranging from normal head shape to turribrachycephaly. Individuals without craniosynostosis have been described [Marzin et al 2020]. Other craniofacial features, such as a prominent forehead, midface retrusion, maxillary hypoplasia, prognathism, and proptosis, are evident at birth. Widely spaced eyes, low-set ears, short nose, anteverted nares, high palate, and gingival overgrowth are other early features. Choanal atresia or nasal obstruction contributes to feeding difficulties, airway and breathing problems, and poor weight gain during infancy and may rarely lead to death [Santos et al 1988]. Craniofacial abnormalities such as midface retrusion and maxillary hypoplasia can contribute to multilevel airway obstruction and may result in obstructive sleep apnea.
Delayed primary and secondary teeth eruption is a common feature during childhood. Skull radiographs typically show impacted permanent tooth buds, with a characteristic copper beaten appearance that may regress by adulthood [Kuthiroly et al 2017] (). Giant cell granuloma of the jaw has also been reported.
Endocrine. Fibroblast growth factor 23 (FGF23) serum levels may increase over time, leading to renal phosphate wasting. Decreased phosphate absorption in the kidneys can result in hypophosphatemia, decreased bone mineralization (with increased risk of fractures), and disturbed vitamin D metabolism. Phosphorus supplementation can be beneficial in those with elevated FGF23-mediated hypophosphatemia.
Neurodevelopment. Development, particularly speech development, can be delayed in early childhood but improves with age. Intelligence is normal unless hydrocephalus or other central nervous system complications occur. For children with multisuture craniosynostosis, early and aggressive surgical intervention to address increased intracranial pressure may prevent intellectual disability. Motor limitation can manifest during childhood as a result of severe cystic bone lesions and osteopenia, leading to bone pain, bone fractures, and skeletal deformities [Kumar et al 2021; A Othman, H Babcock, & C Ferreira, personal observations].
Gastrointestinal. Inguinal hernia and pyloric stenosis have each been reported in two individuals [Kelley et al 1983, Beighton 1989, Holder et al 2017].
Temperature / heat intolerance. Increased body temperature and sensitivity to heat accompanied by excessive sweating has been reported in two individuals [A Othman, H Babcock, & C Ferreira, personal observations].