15q24 Microdeletion Syndrome – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY
Heather Mefford, MD, PhD, Natasha Shur, MD, and Jill Rosenfeld, MS.
Author Information and AffiliationsInitial Posting: February 23, 2012.
Estimated reading time: 17 minutes
Summary
NOTE: THIS PUBLICATION HAS BEEN RETIRED. THIS ARCHIVAL VERSION IS FOR HISTORICAL REFERENCE ONLY, AND THE INFORMATION MAY BE OUT OF DATE.
Clinical characteristics.
The 15q24 microdeletion syndrome is characterized by global developmental delay; mild to severe (usually at least moderate) intellectual disability; facial dysmorphisms; congenital malformations of the hands and feet, eye, and genitalia; joint laxity; and growth retardation and failure to thrive. Less common findings include: seizures; conductive and sensorineural hearing loss; hypospadias and/ or micropenis. Males and females are affected equally.
Diagnosis/testing.
The diagnosis is established by demonstration of a heterozygous deletion at chromosome 15q24, most often involving a 1.1-Mb region between 72.2 and 73.3 Mb of the reference genome (NCBI Build 36 / hg18) using whole-genome and targeted molecular methods that determine the copy number of sequences within the deleted region.
Management.
Treatment of manifestations: Speech, occupational, and physical therapies; routine treatment of ophthalmologic, cardiac, neurologic findings; specialized learning programs to meet individual needs
Surveillance: Routine pediatric care; routine developmental assessments; monitoring of specific identified medical issues
Genetic counseling.
The 15q24 microdeletion syndrome is inherited in an autosomal dominant manner; however, all known cases have resulted from a de novo deletion. Recurrence risk for future pregnancies is low (probably <1%) but greater than that of the general population because of the theoretic possibilities of parental germline mosaicism or a balanced chromosomal rearrangement involving the 15q24 region. Prenatal testing is technically feasible using molecular methods that determine the copy number of sequences within the deleted region.
Diagnosis
Clinical Diagnosis
The clinical spectrum of the 15q24 microdeletion syndrome is variable. Developmental delay and intellectual disability are the most consistent features; however, no single clinical feature is required to establish the diagnosis.
Features that should prompt consideration of this diagnosis in an individual with developmental delay or intellectual disability include:
Dysmorphic facial features, especially a high anterior hairline, deep-set eyes, and a triangular shaped face (see )
Markedly delayed or absent speech
Hypotonia
Joint laxity
Ocular abnormalities, especially strabismus
Hand and foot abnormalities: short fifth fingers; significant shortening of the fourth metacarpals and short fifth metacarpals; thumb anomalies such as proximally implanted thumbs
Growth retardation and failure to thrive
Hearing: conductive and sensorineural hearing loss
Genital anomalies: hypospadias or micropenis in males, labial adhesions in females
Eight individuals with deletions in the 15q24 region. Those in panels A-F each have a deletion that includes the 1.1-Mb critical region. The individual in panel G has a large atypical deletion that includes only the proximal ~250 kb of the critical region; (more...)
Testing
Cytogenetic testing. The 15q24 microdeletion cannot be identified by routine analysis of G-banded chromosomes or other conventional cytogenetic banding techniques.
Molecular Genetic Testing
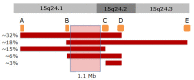
Critical region. The diagnosis of the 15q24 microdeletion is established by demonstration of a heterozygous deletion at chromosome 15q24. The majority of 15q24 deletions identified to date involve a 1.1-Mb region between 72.2-73.3 Mb of the reference genome (NCBI Build 36 / hg18) (identified as breakpoints B and C in ). The actual size and breakpoints of the deletion vary among patients, with most deletions occurring due to nonallelic homologous recombination (NAHR) between segmental duplication blocks.
A schematic representation of the structure of the 15q24 region and the most common deletions identified in the region. Orange rectangles represent segmental duplication blocks A-E, which are though to facilitate nonallelic homologous recombination (NAHR). (more...)
Notably, the critical region continues to be refined as patients with “atypical” deletions are identified, some of which involve only part or none of the proposed 1.1-Mb critical region but still appear to be pathogenic.
Two patients with large deletions that involve only 800 kb [
Andrieux et al 2009] and 500 kb [
Ng et al 2011] of the region between breakpoints B and C in have also been reported. The deletions in these two patients suggest that the
critical region may be smaller than 1.1 Mb, though the large size of their deletions makes it difficult to know which genes are contributing to their features.
Future identification of smaller, atypical deletions will continue to refine the critical region and genotype-phenotype correlations.
Genes. The cognitive features of the 15q24 microdeletion syndrome, including developmental delays and severe speech problems, largely result from deletion of genes in a 1.1-Mb critical region. No single gene in which pathogenic variants cause a similar phenotype has been identified within this region.
Deletion/duplication analysis. The 15q24 microdeletion can be detected by any number of molecular methods that determine the copy number of sequences within the deleted region. Both whole-genome and targeted approaches can be applied.
Genomic microarray technologies. Array-based
genomic hybridization can detect the common
deletion in a
proband. The ability to size the deletion depends on the type of microarray used and the density of probes in the 15q24 region.
Targeted deletion analysis. Targeted methods, including fluorescence in situ hybridization (
FISH), multiplex ligation-dependent probe amplification (MLPA), and
quantitative PCR (qPCR) can be used if the deletion is suspected clinically or for confirmation of the deletion after
genomic microarray analysis. Targeted approaches can also be used to evaluate relatives of the
proband for presence of the deletion.
Whether or not it is possible to size the
deletion depends on the number and distribution of probes in the 15q24 region. It is not possible to size the deletion routinely by use of
FISH.
Table 1.
Molecular Genetic Testing Used in 15q24 Microdeletion
View in own window
| Chromosome Region | Test Method | Variants Detected | Variant Detection Frequency by Test Method 1 |
|---|
| 15q24 | Deletion/duplication analysis 2 | Deletion of 1.1-Mb critical region | ~100% with appropriate probes |
- 1.
The ability of the test method used to detect a deletion or duplication that is present in the indicated chromosomal region
- 2.
Interpretation of test results. Depending on the initial test that identifies the deletion, validation of the deletion by an independent method might be warranted. If high-density genomic microarray platforms have been used for the identification of the deletion, validation of the deletion may not be necessary, as it is unlikely that more than 50-100 adjacent targets show an abnormal copy number by chance.
Testing Strategy
Establishing the diagnosis in a proband requires detection of the 1.1-Mb minimal critical deletion common in 15q24 microdeletion syndrome.
Most microdeletions are detected by
genomic microarray analysis performed as part of the evaluation of developmental delay or intellectual disability.
If the 15q24 microdeletion is suspected based on the clinical features, a targeted technique (e.g.,
FISH, MLPA) can be employed.
Note: The deletion cannot be identified by routine chromosome analysis.
Prenatal diagnosis and preimplantation genetic diagnosis (PGD) for at-risk pregnancies require prior identification of the deletion in the proband and/or of balanced carrier status in a parent. At this point, all known cases have originated de novo; parents with balanced rearrangements have not yet been reported, nor have offspring of individuals with a 15q24 microdeletion who are mildly affected. However, because only a limited group of patients has been studied, a balanced rearrangement in a parent is still a possibility.
Clinical Characteristics
Clinical Description
The 15q24 microdeletion syndrome has a clinically recognizable phenotype that includes developmental delay/intellectual disability, facial dysmorphisms (), congenital malformations, and growth retardation (Table 2). Males and females are affected equally.
Intellectual disability and developmental delay. All persons with a deletion involving the 1.1-Mb critical region reported to date have had mild to severe intellectual disability (ID). Delays are generally global, including gross motor skills, speech, and cognition. Motor delay, most likely related to the degree of hypotonia, is mild to moderate in most. Speech and cognitive deficits are generally more severe. Most affected individuals have severe language delays with limited language acquisition. Cognitive abilities range from mild to severe ID; most have at least moderate ID.
Dysmorphic facial features are common in persons with 15q24 microdeletion syndrome and include high anterior hairline, deep-set eyes, and triangular shaped face. Other frequently reported features include long or prominent philtrum, full lower lip, epicanthal folds, and pointed chin.
Ocular abnormalities have been reported in 59% of individuals, with strabismus being the most frequent. Other rare abnormalities include iris coloboma, chorioretinal coloboma anisocoria, and hypermetropia.
Digital anomalies have been reported in 59% of individuals. These include short fifth fingers, significant shortening of the fourth metacarpals and short fifth metacarpals, thumb anomalies including proximally implanted thumbs, camptodactyly, hypoplastic fifth toes, and toe syndactyly.
Genital anomalies are reported in approximately 30% of patients, with hypospadias and micropenis most commonly reported.
Table 2.
Features of 15q24 Microdeletion Syndrome
View in own window
| Frequency in % | Features |
|---|
>75% of
individuals |
|
| 50%-75% |
|
| 25%-50% | Joint laxity Hearing loss Abnormalities on brain MRI incl heterotopia, corpus callosum cysts, enlarged ventricles, cerebral atrophy, hypoplastic olfactory bulbs Hypospadias &/or micropenis
|
| 10%-25% |
|
| <10% | Heart defects Diaphragmatic hernia Bowel atresia Pierre Robin syndrome
|
Data from 32 individuals (24 male, 8 female) [Sharp et al 2007, Klopocki et al 2008, Marshall et al 2008, El-Hattab et al 2009, Masurel-Paulet et al 2009, Van Esch et al 2009, El-Hattab et al 2010, McInnes et al 2010, Ng et al 2011, Mefford et al 2012]
Penetrance
Penetrance is 100%: clinical features of 15q24 microdeletion syndrome are apparent in all individuals with the microdeletion, although the extent and severity of clinical findings vary among individuals.
Prevalence
The exact prevalence of the 15q24 microdeletion syndrome is unknown; though it is clearly rare. To date, about 30 affected individuals have been reported worldwide.
Large-scale surveys of the novel 15q24 microdeletion syndrome estimate the frequency to be 0.1-0.2% in individuals with autism spectrum disorders [McInnes et al 2010].
In one large series, 15q24 microdeletions were identified in 3:10,000-4:10,000 individuals in clinical array CGH studies [Mefford et al 2012].
Syndromic microphthalmia / Matthew Wood syndrome (recessive). Recessive pathogenic variants in STRA6 cause a profound intellectual disability syndrome with eye abnormalities including microphthalmia and/or anophthalmia, diaphragmatic hernia, pulmonary hypoplasia, cardiac defects, and short stature [Golzio et al 2007, Pasutto et al 2007].
Congenital adrenal insufficiency (recessive) (OMIM 118485). Compound heterozygous or homozygous pathogenic variants in CYP11A1 have been reported in several persons with congenital adrenal insufficiency with partial or complete 46,XY sex reversal [Katsumata et al 2002, Hiort et al 2005, Al Kandari et al 2006, Kim et al 2008, Rubtsov et al 2009]. One individual in whom only one pathogenic variant in the gene was identified has also been reported [Tajima et al 2001].
MPI-CDG (CDG-Ib; recessive) (OMIM 602579). This autosomal recessive disorder is caused by compound heterozygous or homozygous pathogenic variants in MPI, located within the 15q24 microdeletion critical region [Jaeken et al 1998, Schollen et al 2000]. Features of MPI-CDG include chronic diarrhea, failure to thrive, protein-losing enteropathy, and coagulopathy. Unlike other types of CDG, MPI-CDG does not usually involve the central nervous system. (See Congenital Disorders of Glycosylation Overview.)
Reciprocal duplication. Several individuals have been reported to have duplications of the 15q24 region that include the 1.1-Mb critical region for the 15q24 microdeletion syndrome. It is currently unclear if the 15q24 duplications represent a distinct clinical phenotype [El-Hattab et al 2010]. Two reported, unrelated individuals share some features similar to the 15q24 microdeletion syndrome, including developmental delay, digital anomalies, and dysmorphic features [Kiholm Lund et al 2008, El-Hattab et al 2009]. In contrast, the only feature shared with two additional individuals, who were more severely affected and also had secondary copy number changes, was developmental delay [El-Hattab et al 2010]. In the three instances in which inheritance was known, the duplications were inherited from mildly affected or healthy parents. Given the parental phenotypes and the finding of secondary copy number changes in some affected individuals, it is possible that these duplications are one of several factors contributing to an abnormal phenotype, or show reduced penetrance and/or variable expressivity.
15q24 duplications of the region distal to the 1.1-Mb critical deletion region have also been reported and may also contribute to variable abnormal phenotypes. Two reported families have multiple affected individuals with this duplication; persons heterozygous for this duplication showed varying degrees of developmental delay, hypotonia, and dysmorphic features; one of the families also had individuals with autism spectrum disorders [El-Hattab et al 2009, Roetzer et al 2010].
Differential Diagnosis
The most common findings in 15q24 microdeletion syndrome, developmental delay and childhood hypotonia, are frequent and relatively nonspecific indications for molecular cytogenetic analysis. However, the concurrent finding of characteristic facial dysmorphic features and hand and genital anomalies may prompt special consideration of 15q24 microdeletion syndrome.
Other diagnoses that may be considered in affected individuals include the following:
Angelman syndrome. Clinical features seen in 15q24
microdeletion syndrome that may also be seen in Angelman syndrome include microcephaly, absent speech, growth retardation, seizures, and generally happy disposition.
Fryns syndrome is characterized by diaphragmatic defects (diaphragmatic hernia, eventration, hypoplasia or agenesis); characteristic facial appearance (coarse facies, ocular hypertelorism, broad and flat nasal bridge, thick nasal tip, long philtrum, low-set and poorly formed ears, tented upper lip, macrostomia, micrognathia); distal digital hypoplasia (nails, terminal phalanges); pulmonary hypoplasia; and associated anomalies (polyhydramnios, cloudy corneas and/or microphthalmia, orofacial clefting, renal dysplasia/renal cortical cysts, and/or malformation involving brain, cardiovascular system, gastrointestinal system, genitalia). Survival beyond the neonatal period has been rare. Data on postnatal growth and psychomotor development are limited; however, severe developmental delay and intellectual disability are common. Since persons with 15q24
microdeletion syndrome have been reported to have diaphragmatic hernia, Fryns syndrome should be considered in the differential diagnosis.
Prader-Willi syndrome
(PWS). Clinical features of 15q24
microdeletion syndrome that may also be seen with PWS include severe neonatal hypotonia, seizures, global developmental delay, strabismus, upslanting palpebral fissures, and cryptorchidism. However, in contrast to PWS, childhood hyperphagia and central obesity have not been reported in 15q24 microdeletion syndrome, and behavioral problems and sleep disturbances are less common.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with the 15q24 microdeletion syndrome, the following evaluations should be considered:
Ophthalmologic examination
Formal audiology evaluation to assess for sensorineural hearing loss
Cardiac evaluation
Brain imaging in individuals with microcephaly and/or neurologic findings
Examination for genitourinary abnormalities (e.g., hypospadias or cryptorchidism in males)
Comprehensive developmental assessment
Consideration of neurologic referral for any unusual movements or concern for seizures
Discussion of results with a clinical geneticist or genetic counselor
Treatment of Manifestations
The following are indicated:
Routine treatment of ophthalmologic, cardiac, neurologic findings
Speech, occupational, and physical therapies
Specialized learning programs to meet individual needs
No specific antiepileptic or antipsychotic medications are indicated
Surveillance
Appropriate surveillance includes:
Pregnancy Management
If a fetus is known to have 15q24 microdeletion syndrome, fetal echocardiogram and ultrasound examination with an attempt to visualize the palate and look for diaphragmatic hernia are recommended. Close monitoring for intrauterine growth retardation is warranted. Counseling with regard to the developmental outcomes and medical complications of the 15q24 microdeletion syndrome is appropriate. Delivery at a center with a good neonatal intensive care team is optimal, as respiratory complications and feeding difficulties may occur after delivery.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and www.ClinicalTrialsRegister.eu in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Risk to Family Members
Parents of a proband
Sibs of a proband
The risk to the sibs of the
proband depends on the status of the parents. In the unlikely event that a parent has
germline mosaicism for a 15q24
deletion or a balanced structural
chromosome rearrangement involving the 15q24 region, the risk to sibs is increased and depends on the specific chromosome rearrangement.
When the parents are clinically unaffected, the risk to the sibs of a
proband appears to be low because recurrence has not yet been reported in a family with the 15q24
microdeletion syndrome.
Offspring of a proband. No individuals diagnosed with 15q24 microdeletion syndrome have been known to reproduce; however, information on adults with 15q24 microdeletion syndrome is limited. Individuals who have the 15q24 deletion syndrome would be expected to have a 50% chance of transmitting the deletion to each child.
Other family members. Families in whom more than one individual is affected have not been reported to date.
Prenatal Testing and Preimplantation Genetic Diagnosis
Prenatal testing may be offered to unaffected parents who have had a child with the 15q24 microdeletion syndrome because of the recurrence risk (probably <1%) associated with the possibility of germline mosaicism.
Prenatal testing is technically feasible. Fetal cells obtained by amniocentesis usually performed at approximately 15 to 18 weeks' gestation or CVS at approximately ten to 12 weeks' gestation can be analyzed using array comparative genomic hybridization or targeted deletion analysis methods in the manner described in Molecular Genetic Testing.
Note: Gestational age is expressed as menstrual weeks calculated either from the first day of the last normal menstrual period or by ultrasound measurements.
Preimplantation genetic diagnosis (PGD) may be an option for some families in which the deletion has been identified.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
My46Trait Profile
Chromosome Disorder Outreach (CDO)
PO Box 724
Boca Raton FL 33429-0724
Phone: 561-395-4252 (Family Helpline)
Email: info@chromodisorder.org
Unique: The Rare Chromosome Disorder Support Group
G1 The Stables
Station Road West
Oxted Surrey RH8 9EE
United Kingdom
Phone: +44 (0) 1883 723356
Email: info@rarechromo.org; rarechromo@aol.com
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
15q24 Microdeletion: Genes and Databases
View in own window
| Gene | Chromosome Locus | Protein | ClinVar |
|---|
| Unknown | 15q24 | Unknown | |
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Molecular Genetic Pathogenesis
The 15q24 region is characterized by several segmental duplication (SD) blocks or low copy repeats [Bailey et al 2002, El-Hattab et al 2009] (see ). The SD blocks are thought to facilitate nonallelic homologous recombination (NAHR), resulting in microdeletion or duplication of the intervening region. Five SD blocks (A, B, C, D, E) have been identified as playing a role in generating recurrent 15q24 microdeletions, with the most common deletions occurring between blocks A and D (3.1 Mb), B and E (3.8 Mb), and A and C (2.6 Mb). The critical region, which lies between B and C, was identified as the smallest region of overlap among the common deletions; no one with a deletion involving only the critical region has been reported to date.
The 1.1-Mb critical region between blocks B and C contains 26 genes that are well characterized in the RefSeq database. Haploinsufficiency of one or more genes in the region is thought to result in the 15q24 microdeletion syndrome phenotype; however, no pathogenic variants in a single gene from the region have yet been identified as causing a similar phenotype. Candidate genes, based on their known function, include the following:
CPLX3, which is expressed in the brain and eye, regulates neurotransmitter release in mouse hippocampal neurons, and is involved in syntaxin binding;
SEMA7A, a membrane-anchored semaphorin that mediates central and peripheral axon growth and is required for proper axon tract formation during development.
References
Literature Cited
al Kandari H, Katsumata N, Alexander S, Rasoul MA. Homozygous mutation of P450 side-chain cleavage enzyme gene (CYP11A1) in 46,XY patient with adrenal insufficiency, complete sex reversal, and agenesis of corpus callosum.
J Clin Endocr Metab. 2006;91:2821–6. [
PubMed: 16705068]
Andrieux J, Dubourg C, Rio M, Attie-Bitach T, Delaby E, Mathieu M, Journel H, Copin H, Blondeel E, Doco-Fenzy M, Landais E, Delobel B, Odent S, Manouvrier-Hanu S, Holder-Espinasse M. Genotype-phenotype correlation in four 15q24 deleted patients identified by array-CGH.
Am J Med Genet A. 2009;149A:2813–9. [
PMC free article: PMC2874573] [
PubMed: 19921647]
Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome.
Science. 2002;297:1003–7. [
PubMed: 12169732]
El-Hattab AW, Smolarek TA, Walker ME, Schorry EK, Immken LL, Patel G, Abbott MA, Lanpher BC, Ou Z, Kang SH, Patel A, Scaglia F, Lupski JR, Cheung SW, Stankiewicz P. Redefined genomic architecture in 15q24 directed by patient deletion/duplication breakpoint mapping.
Hum Genet. 2009;126:589–602. [
PMC free article: PMC3669685] [
PubMed: 19557438]
El-Hattab AW, Zhang F, Maxim R, Christensen KM, Ward JC, Hines-Dowell S, Scaglia F, Lupski JR, Cheung SW. Deletion and duplication of 15q24: molecular mechanisms and potential modification by additional copy number variants.
Genet Med. 2010;12:573–86. [
PubMed: 20860070]
Golzio C, Martinovic-Bouriel J, Thomas S, Mougou-Zrelli S, Grattagliano-Bessieres B, Bonniere M, Delahaye S, Munnich A, Encha-Razavi F, Lyonnet S, Vekemans M, Attie-Batich T, Etchevers HC. Matthew-Wood syndrome is caused by truncating mutations in the retinol-binding protein receptor gene STRA6.
Am J Hum Genet. 2007;80:1179–87. [
PMC free article: PMC1867105] [
PubMed: 17503335]
Hiort O, Holterhus PM, Werner R, Marschke C, Hoppe U, Partsch CJ, Riepe FG, Achermann JC, Struve D. Homozygous disruption of P450 side-chain cleavage (CYP11A1) is associated with prematurity, complete 46,XY sex reversal, and severe adrenal failure.
J Clin Endocrinol Metab. 2005;90:538–41. [
PubMed: 15507506]
Jaeken J, Matthijs G, Saudubray J-M, Dionisi-Vici C, Bertini E, de Lonlay P, Henri H, Carchon H, Schollen E, Van Schaftingen E. Phosphomannose isomerase deficiency: a carbohydrate-deficient glycoprotein syndrome with hepatic-intestinal presentation.
Am J Hum Genet. 1998;62:1535–9. [
PMC free article: PMC1377152] [
PubMed: 9585601]
Katsumata N, Ohtake M, Hojo T, Ogawa E, Hara T, Sato N, Tanaka T. Compound heterozygous mutations in the cholesterol side-chain cleavage enzyme gene (CYP11A) cause congenital adrenal insufficiency in humans.
J Clin Endocr Metab. 2002;87:3808–13. [
PubMed: 12161514]
Kiholm Lund AB, Hove HD, Kirchhoff M. A. 15q24 microduplication, reciprocal to the recently described 15q24 microdeletion, in a boy sharing clinical features with 15q24 microdeletion syndrome patients.
Eur J Med Genet. 2008;51:520–6. [
PubMed: 18755302]
Kim CJ, Lin L, Huang N, Quigley CA. AvRuskin TW, Achermann JC, Miller WL. Severe combined adrenal and gonadal deficiency caused by novel mutations in the cholesterol side chain cleavage enzyme, P450scc.
J Clin Endocr Metab. 2008;93:696–702. [
PMC free article: PMC2266942] [
PubMed: 18182448]
Klopocki E, Graul-Neumann LM, Grieben U, Tönnies H, Ropers HH, Horn D, Mundlos S, Ullmann R. A further case of the recurrent 15q24 microdeletion syndrome, detected by array CGH.
Eur J Pediatr. 2008;167:903–8. [
PMC free article: PMC2757600] [
PubMed: 17932688]
Ng ISL, Chin WH, Lim ECP, Tan EC. An additional case of the recurrent 15q24.1 microdeletion syndrome and review of the literature.
Twin Res Hum Genet. 2011;14:333–9. [
PubMed: 21787116]
Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, Thiruvahindrapduram B, Fiebig A, Schreiber S, Friedman J, Ketelaars CE, Vos YJ, Ficicioglu C, Kirkpatrick S, Nicolson R, Sloman L, Summers A, Gibbons CA, Teebi A, Chitayat D, Weksberg R, Thompson A, Vardy C, Crosbie V, Luscombe S, Baatjes R, Zwaigenbaum L, Roberts W, Fernandez B, Szatmari P, Scherer SW. Structural variation of chromosomes in autism spectrum disorder.
Am J Hum Genet. 2008;82:477–88. [
PMC free article: PMC2426913] [
PubMed: 18252227]
Masurel-Paulet A, Callier P, Thauvin-Robinet C, Chouchane M, Mejean N, Marle N, Mosca AL, Ben Salem D, Giroud M, Guibaud L, Huet F, Mugneret F, Faivre L. Multiple cysts of the corpus callosum and psychomotor delay in a patient with a 3.1 Mb 15q24.1q24.2 interstitial deletion identified by array-CGH.
Am J Med Genet A. 2009;149A:1504–10. [
PubMed: 19533778]
McInnes LA, Nakamine A, Pilorge M, Brandt T, Jiménez González P, Fallas M, Manghi ER, Edelmann L, Glessner J, Hakonarson H, Betancur C, Buxbaum JD. A large-scale survey of the novel 15q24 microdeletion syndrome in autism spectrum disorders identifies an atypical deletion that narrows the critical region.
Mol Autism. 2010;1:5. [
PMC free article: PMC2907565] [
PubMed: 20678247]
Mefford HC, Rosenfeld JA, Shur N, Slavotinek AM, Cox VA, Hennekam RC, Firth HV, Willatt L, Wheeler P, Morrow EM, Cook J, Sullivan R, Oh A, McDonald MT, Zonana J, Keller K, Hannibal MC, Ball S, Kussmann J, Gorski J, Zelewski S, Banks V, Smith W, Smith R, Paull L, Rosenbaum KN, Amor DJ, Silva J, Lamb A, Eichler EE. Further clinical and molecular delineation of the 15q24 microdeletion syndrome.
J Med Genet. 2012;49:110–8. [
PMC free article: PMC3261729] [
PubMed: 22180641]
Ng ISL, Chin WH, Lim ECP, Tan EC. An additional case of the recurrent 15q24.1 microdeletion syndrome and review of the literature.
Twin Res Hum Genet. 2011;14:333–9. [
PubMed: 21787116]
Pasutto F, Sticht H, Hammersen G, Gillessen-Kaesbach G, Fitzpatrick DR, Nürnberg G, Brasch F, Schirmer-Zimmermann H, Tolmie JL, Chitayat D, Houge G, Fernández-Martínez L, Keating S, Mortier G, Hennekam RC, von der Wense A, Slavotinek A, Meinecke P, Bitoun P, Becker C, Nürnberg P, Reis A, Rauch A. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation.
Am J Hum Genet. 2007;80:550–60. [
PMC free article: PMC1821097] [
PubMed: 17273977]
Roetzer KM, Schwarzbraun T, Obenauf AC, Hauser E, Speicher MR. Further evidence for the pathogenicity of 15q24 microduplications distal to the minimal critical regions.
Am J Med Genet A. 2010;152A:3173–8. [
PubMed: 21108404]
Rubtsov P, Karmanov M, Sverdlova P, Spirin P, Tiulpakov A. A novel homozygous mutation in CYP11A1 gene is associated with late-onset adrenal insufficiency and hypospadias in a 46,XY patient.
J Clin Endocr Metab. 2009;94:936–9. [
PubMed: 19116240]
Schollen E, Dorland L, de Koning TJ, Van Diggelen OP, Huijmans JGM, Marquardt T, Babovic-Vuksanovic D, Patterson M, Imtiaz F, Winchester B, Adamowicz M, Pronicka E, Freeze H, Matthijs G. Genomic organization of the human phosphomannose isomerase (MPI) gene and mutation analysis in patients with congenital disorders of glycosylation type Ib (CDG-Ib).
Hum Mutat. 2000;16:247–52. [
PubMed: 10980531]
Sharp AJ, Selzer RR, Veltman JA, Gimelli S, Gimelli G, Striano P, Coppola A, Regan R, Price SM, Knoers NV, Eis PS, Brunner HG, Hennekam RC, Knight SJ, de Vries BB, Zuffardi O, Eichler EE. Characterization of a recurrent 15q24 microdeletion syndrome.
Hum Mol Genet. 2007;16:567–72. [
PubMed: 17360722]
Tajima T, Fujieda K, Kouda N, Nakae J, Miller WL. Heterozygous mutation in the cholesterol side chain cleavage enzyme (P450scc) gene in a patient with 46,XY sex reversal and adrenal insufficiency.
J Clin Endocr Metab. 2001;86:3820–5. [
PubMed: 11502818]
Van Esch H, Backx L, Pijkels E, Fryns JP. Congenital diaphragmatic hernia is part of the new 15q24 microdeletion syndrome.
Eur J Med Genet. 2009;52:153–6. [
PubMed: 19233321]