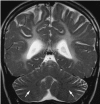
Clinical Description
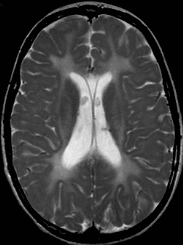
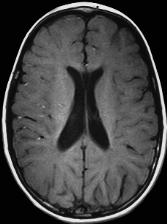
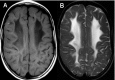
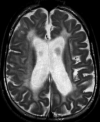
Hypomyelination and congenital cataract (HCC) phenotype is quite consistent in the affected individuals described to date.
Table 2.
Hypomyelination and Congenital Cataract: Frequency of Select Features
View in own window
| Feature | Proportion of Persons w/Feature | Comment |
|---|
| Bilateral congenital cataracts | 26/30 | |
| Developmental delay | 30/30 | |
| Intellectual disability | 30/30 | |
| Dysarthria | 26/26 | |
| Truncal hypotonia | 26/26 | |
| Brisk tendon reflexes & bilateral extensor plantar responses | 30/30 | |
| Cerebellar signs | 11/25 | Truncal titubation, intention tremor |
| Peripheral neuropathy | 22/24 | Muscle weakness, muscle wasting of the legs |
| Seizures | 4/28 | Seizures may be prolonged & w/fever. |
Prenatal/perinatal. All affected individuals have normal prenatal and perinatal histories.
Ophthalmologic. Bilateral congenital cataracts identified at birth or within the first month of life are the first clinical sign. All children underwent ocular surgery in the first months of life with the exception of the one child who had adolescent-onset cataracts [Ugur & Tolun 2008].
Psychomotor development is normal up until the end of the first year of life, when developmental delays appear [Biancheri et al 2007]. The ability to walk with support is achieved between ages 12 and 24 months. Independent walking is not achieved in all individuals. Slowly progressive neurologic impairment then becomes apparent with gradual loss of the ability to walk. Most individuals become wheelchair bound between ages eight and nine years [Biancheri et al 2007].
Feeding issues occur as a result of neurologic impairment. Swallowing may become difficult, and growth may be affected by suboptimal intake.
Cognitive skills. All individuals have mild-to-moderate intellectual disability without deterioration in cognitive ability over time.
Neurologic findings. Clinical examination reveals the following from the onset of the disease course:
Seizures including those triggered by fever may occur, but are not a predominant clinical feature.
Neurophysiologic investigations show the following from the onset of the disease course:
Motor nerve conduction velocity. Slightly to markedly slowed in most individuals, with lower values in older persons
Compound muscle action potentials. Reduced amplitude
Electromyography. Signs of denervation in the absence of spontaneous activity
Waking EEG. Irregular background activity; multifocal epileptiform discharges may be recorded.
Brain stem auditory evoked potentials. Increased I-V interpeak conduction time in individuals older than age two years
Electroretinogram. Normal
Neuropathologic findings
Sural nerve biopsy of individuals with peripheral neuropathy shows a slight-to-severe reduction in density of myelinated fibers, with several axons surrounded by a thin myelin sheath or devoid of myelin.
Uncompaction of the myelin sheath, which in some fibers appears redundant and irregularly folded, is occasionally seen.
Electron microscopy confirms the presence of axons devoid of myelin, together with thinly myelinated fibers, sometimes surrounded by few Schwann cells processes, forming small onion bulbs.
Orthopedic
issues. A slowly progressive scoliosis appears concurrently with the loss of the ability to walk [Biancheri et al 2007].
Life expectancy is unknown; the oldest living affected individual is age 34 years.
Genotype-Phenotype Correlations
Pathogenic variants leading to the complete absence of HYCC1 (formerly FAM126A) protein expression are associated with the full phenotype of bilateral cataract, central nervous system hypomyelination, and peripheral nerve hypomyelination.
Pathogenic variants leading to a partial protein deficiency are associated with the milder form without peripheral nervous system involvement.
An individual with deletion of exons 8 and 9 did not have congenital cataracts; cataracts developed at age nine years. A second individual had congenital unilateral cataract. However, of the four children in this family who survived beyond age two years, none was able to walk even with support after age six years [Ugur & Tolun 2008].
Because of the limited number of individuals with HCC described so far, these correlations should be further confirmed.