Clinical Description
Neuralgic amyotrophy attacks. Typically, onset of painful attacks in hereditary neuralgic amyotrophy (HNA) occurs in the second or third decade of life (median age of onset 28 years), but children as young as age one year have had attacks. The male to female ratio is 2:1.
The attacks comprise severe aching, burning, or stabbing pains, most often in the shoulders, neck, and/or arm region, followed by multifocal atrophy and paresis. Usually the brachial plexus is involved. In one third of cases, the involvement is bilateral, although severity is usually asymmetric. Attacks appear to become less frequent with age.
The most comprehensive review of attack features in both HNA and sporadic idiopathic neuralgic amyotrophy (see Differential Diagnosis) was reported by van Alfen & van Engelen [2006]. The pain lasts an average of four weeks. Weakness most often begins in the periscapular or perihumeral muscles (see ) between one and two weeks after the onset of pain. In some instances the onset of weakness may follow within 24 hours of the onset of pain.
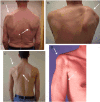
Different presentations of upper-extremity atrophy and paresis A. On the left: atrophy of supraspinatus and infraspinatus muscles and rhomboid muscles (white arrow); on the right: scapular tilting and rotation caused by serratus anterior muscle weakness (more...)
The long thoracic and suprascapular nerves are affected in about 70% of cases. Other frequently involved nerves are the axillary, musculocutanous, radial, and anterior interosseus. Lower plexus involvement (median motor and ulnar distribution) occurs in about 5% [van Alfen 2011].
In many cases the muscle weakness may go unnoticed, especially if it only affects the periscapular muscles such as the serratus anterior, rhomboids or subscapularis. Functionally, however, the resulting scapular instability often causes pain, limitation of movement, and exercise intolerance of the affected limb that can persist for months to years.
Sensory symptoms, present in the majority of affected individuals, are often overlooked. They can include the following:
While the shoulder and arm are primarily affected by attacks in HNA, other sites that may also be involved in an attack include the following:
Lumbosacral plexus in ~33% of attacks
Phrenic nerve palsy in 14% of attacks; may cause orthopnea, respiratory distress and sleep disturbance
Recurrent laryngeal nerve in 3% of attacks; may cause vocal cord paresis resulting in hoarseness and hypophonia
Facial nerve or other cranial nerves (rarely)
Two patterns of HNA attacks are described:
The prognosis for eventual recovery of neurologic function in neuralgic amyotrophy is guarded, with residual deficits accumulating with additional attacks.
Characteristic physical features. In some families, HNA is associated with non-neurologic physical features that allow assessment of the risk for HNA before attacks appear. Typically, these non-neurologic findings include short stature; partial syndactyly of the fingers or toes; characteristic craniofacial features with relatively closely spaced eyes, short palpebral fissures, and epicanthus; and cleft (bifid) uvula or cleft palate [Jeannet et al 2001]. The ocular hypotelorism in some families is striking, with interpupillary distance typically between -1 and -2 standard deviations. As pointed out by several authors, the facial features of persons with HNA resemble portraits painted by the artist Amedeo Modigliani [Dunn et al 1978].
Excessive partial circumferential skin folds of the neck and arms are also characteristic features [Jeannet et al 2001].
Pathophysiology. Attacks may be triggered by periods of physical, immunologic, or emotional stress. Females appear to have a predilection for attacks after childbirth. This, and association of attacks following immunizations and recent viral or bacterial infections, raise a possible role of an immune system trigger. Prior strenuous usage of the upper limbs has also been reported to precipitate attacks suggesting that local trauma or ischemia of the brachial plexus resulting from compression between muscle groups may underlay the plexopathy, making it more susceptible to (auto-) immune damage.
Biopsy of sural or superficial radial nerves is rarely performed in this disorder. The only finding described in the majority of biopsies is focal decreases in myelinated fibers within individual nerve fascicles [van Alfen et al 2005]. In one report, multiple epineural perivascular mononuclear infiltrates without necrosis were seen in three of four upper-extremity nerve biopsies, obtained three weeks, three months, and seven months after onset of an attack [Klein et al 2002]. These infiltrates were accompanied by active axonal degeneration.
Prevalence
The prevalence of HNA is unknown. About 300 families are known worldwide.
The prevalence of HNA is estimated to be about an order of magnitude less than that of idiopathic neuralgic amyotrophy (Parsonage-Turner syndrome), which has an estimated incidence of 1.64:100,000/year to 3:100,000/year [Beghi et al 1985, MacDonald et al 2000].
Prevalence of any brachial neuritis was estimated to be 3:10,000 in the London (UK) area [MacDonald et al 2000].
The actual prevalence of these disorders is likely to be higher because of underdiagnosis. Sixty percent of individuals with neuralgic amyotrophy seen at the Nijmegen clinical center were first diagnosed with a different disorder [van Alfen & van Engelen 2006].