Summary
Clinical characteristics.
Citrin deficiency can manifest in newborns or infants as neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD), in older children as failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD), and in adults as recurrent hyperammonemia with neuropsychiatric symptoms in citrullinemia type II (CTLN2). Often citrin deficiency is characterized by strong preference for protein-rich and/or lipid-rich foods and aversion to carbohydrate-rich foods.
NICCD. Children younger than age one year have a history of low birth weight with growth restriction and transient intrahepatic cholestasis, hepatomegaly, diffuse fatty liver, and parenchymal cellular infiltration associated with hepatic fibrosis, variable liver dysfunction, hypoproteinemia, decreased coagulation factors, hemolytic anemia, and/or hypoglycemia. NICCD is generally not severe and symptoms often resolve by age one year with appropriate treatment, although liver transplantation has been required in rare instances.
FTTDCD. Beyond age one year, many children with citrin deficiency develop a protein-rich and/or lipid-rich food preference and aversion to carbohydrate-rich foods. Clinical abnormalities may include growth restriction, hypoglycemia, pancreatitis, severe fatigue, anorexia, and impaired quality of life. Laboratory changes are dyslipidemia, increased lactate-to-pyruvate ratio, higher levels of urinary oxidative stress markers, and considerable deviation in tricarboxylic acid (TCA) cycle metabolites. One or more decades later, some individuals with NICCD or FTTDCD develop CTLN2.
CTLN2. Presentation is sudden and usually between ages 20 and 50 years. Manifestations are recurrent hyperammonemia with neuropsychiatric symptoms including nocturnal delirium, aggression, irritability, hyperactivity, delusions, disorientation, restlessness, drowsiness, loss of memory, flapping tremor, convulsive seizures, and coma. Symptoms are often provoked by alcohol and sugar intake, medication, and/or surgery. Affected individuals may or may not have a prior history of NICCD or FTTDCD.
Diagnosis/testing.
The diagnosis of citrin deficiency is established in an individual with characteristic biochemical findings (in general, increased blood or plasma concentration of ammonia, plasma or serum concentration of citrulline and arginine, plasma or serum threonine-to-serine ratio, and serum concentration of pancreatic secretory trypsin inhibitor) and identification of biallelic pathogenic variants in SLC25A13.
Management.
Treatment of manifestations: NICCD: Supplement diet with fat-soluble vitamins and use of lactose-free and medium-chain triglyceride (MCT)-enriched formula. FTTDCD: In addition to dietary treatment, administration of sodium pyruvate may improve growth. CTLN2: Liver transplantation prevents hyperammonemic crises, corrects metabolic disturbances, and eliminates preferences for protein-rich foods; arginine administration decreases blood ammonia concentration and reduced calorie/carbohydrate intake; increased protein intake lessens hypertriglyceridemia. Use of arginine, sodium pyruvate, and MCT oil may delay the need for liver transplantation.
Prevention of primary manifestations: Lipid and protein-rich low-carbohydrate diet.
Surveillance: Periodic measurement of plasma concentration of ammonia and citrulline, and serum concentration of PSTI for all phenotypes associated with citrin deficiency. Follow up of children who have had NICCD for the laboratory and physical findings of FTTDCD.
Agents/circumstances to avoid: Low-protein high-carbohydrate diets; glycerol and fructose infusions for brain edema; alcohol; acetaminophen and rabeprozole.
Evaluation of relatives at risk: It is appropriate to identify affected sibs of a proband so that appropriate dietary management can be instituted before symptoms occur.
Genetic counseling.
Citrin deficiency is inherited in an autosomal recessive manner. When both parents are carriers of an SLC25A13 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. When one parent is a carrier and the other parent has two SLC25A13 pathogenic variants, each sib of an affected individual has at conception a 50% chance of being affected and a 50% chance of being an asymptomatic carrier. Testing for at-risk relatives and prenatal testing for a pregnancy at increased risk are possible if the SLC25A13 pathogenic variants in the family are known.
GeneReview Scope
View in own window
| Citrin Deficiency: Included Phenotypes 1 |
|---|
Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) Failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD) Citrullinemia type II (CTLN2)
|
Diagnosis
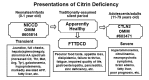
Citrin deficiency has two distinct well-recognized phenotypes: neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) and citrullinemia type II (CTLN2) (see ) [Saheki & Kobayashi 2002, Yamaguchi et al 2002, Kobayashi & Saheki 2004, Saheki & Kobayashi 2005, Kobayashi et al 2006] – and a third intermediate phenotype: failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD) [Song et al 2011, Song et al 2013].
Clinical and laboratory manifestations of citrin deficiency AA = amino acids; AFP = αlpha-fetoprotein; Arg = arginine; Cit = citrulline; CTLN2 = adult-onset type II citrullinemia; FTTDCD = failure to thrive and dyslipidemia caused by citrin deficiency; (more...)
Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) is characterized by transient neonatal cholestasis and variable hepatic dysfunction. Some affected individuals have a poor prognosis due to liver cirrhosis.
Failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD) is characterized by post-NICCD growth restriction before CTLN2 onset and abnormalities of serum lipid concentrations including triglycerides, total cholesterol, and HDL-cholesterol. Clinical diagnosis of citrin deficiency during this stage is difficult in the absence of a history of unique food preferences or without molecular testing.
Citrullinemia type II (CTLN2) is characterized by childhood- to adult-onset recurring episodes of hyperammonemia and associated neuropsychiatric symptoms.
Suggestive Findings
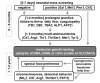
Citrin deficiency should be suspected in individuals with the following findings (see ):
Flow chart for diagnosis of citrin deficiency Note that food preferences (e.g., aversion to sugars) are important in the diagnosis of citrin deficiency, not only in typical CTLN2 but also in cases of growth restriction, hypoglycemia, pancreatitis, and (more...)
Infants who have had a positive newborn screening test for:
Citrullinemia and/or prolonged jaundice; or
Galactosemia, hypermethionemia, tyrosinemia or hyperphenylalanemia, who on follow-up diagnostic testing were found not to have one of these disorders.
Note: Plasma concentration of galactose, methionine, and/or phenylalanine is elevated in newborn screening blood spots in approximately 40% of children with NICCD [
Ohura et al 2003,
Ohura et al 2007]. Although plasma tyrosine is elevated in some individuals, this is not a common feature. Instead, tyrosinemia is suspected because the 4-hydroxyphenyllactate and 4-hydroxyphenylpyruvate are increased on urinary GC-MS analysis.
Children beyond age one year who present with failure to thrive and dyslipidemia
Older children and adults with hepatic encephalopathy with hyperammonemia, especially those with a history of aversion to carbohydrate and fondness for protein- and lipid-rich foods
Children and adults with unexplained recurrent pancreatitis, hyperlipidemia, fatty liver, or hepatoma
Establishing the Diagnosis
The diagnosis of citrin deficiency is established in a proband with biochemical testing results consistent with citrin deficiency and/or biallelic pathogenic variants in SLC25A13 identified on molecular genetic testing (see Table 3). Western blot analysis is available if the pathogenic variants cannot be identified.
Biochemical Testing
The diagnosis of citrin deficiency is further supported by results of the following testing as indicated in Table 1:
Table 1.
Biochemical Findings in Citrin Deficiency by Phenotype
View in own window
| Phenotype (Age) | Blood or Plasma Concentration of Ammonia (µmol/L) | Plasma or Serum Concentration of: | Plasma or Serum Threonine-to-Serine Ratio | Serum Concentration of PSTI 2 (ng/mL) |
|---|
| Citrulline 1 (µmol/L) | Arginine (µmol/L) |
|---|
| Control | 18-47 3 | 17-43 3 | 54-130 3 | 1.10 | 4.6-20 3 |
NICCD
(0-6 mos) | 60 | 300 | 205 | 2.29 | 30 |
FTTDCD
(>1-11 yrs) | Normal or slightly elevated | Normal or slightly elevated | Usually normal | Unknown | Unknown |
CTLN2
(11-79 yrs) | 152 | 418 | 198 | 2.32 | 71 |
PSTI = pancreatic secretory trypsin inhibitor
- 1.
Citrullinemia, which can be detected on newborn screening, is the earliest identifiable biochemical abnormality of NICCD [Tamamori et al 2004].
- 2.
Because the serum PSTI concentration is high in some individuals with NICCD [Tamamori et al 2002] and also in individuals before the onset of CTLN2 [Tsuboi et al 2001], the measurement of serum PSTI concentration may be useful in presymptomatic diagnosis of CTLN2.
- 3.
Table 2.
Plasma Concentrations of Threonine, Methionine, and Tyrosine at Age 0-6 Months in NICCD
View in own window
| Amino Acid | Median
(25%-75% Range) (µmol/L) | Control Range
(µmol/L) |
|---|
| Threonine | 496 (291-741) | 67-190 |
| Methionine | 124 (53-337) | 19-40 |
| Tyrosine | 178 (99-275) | 40-90 |
Molecular Genetic Testing
Molecular genetic testing approaches can include single-gene testing and use of a multigene panel.
Single-gene testing. Sequence analysis of SLC25A13 is performed first and followed by gene-targeted deletion/duplication analysis if only one or no pathogenic variant is found.
Targeted analysis for pathogenic variants can be performed first in individuals of Japanese or Chinese ancestry.
In Japanese persons with citrin deficiency, two pathogenic variants account for the majority (~70%) of pathogenic alleles [Authors, personal experience] (see
Molecular Genetics,
Pathogenic variants).
In Chinese persons with citrin deficiency, four pathogenic variants account for more than 80% of the pathogenic alleles [
Lin et al 2016] (see
Molecular Genetics,
Pathogenic variants).
A multigene panel that includes SLC25A13 and other genes of interest (see Differential Diagnosis) may be considered. Note: (1) The genes included in the panel and the diagnostic sensitivity of the testing used for each gene vary by laboratory and are likely to change over time. (2) Some multigene panels may include genes not associated with the condition discussed in this GeneReview; thus, clinicians need to determine which multigene panel is most likely to identify the genetic cause of the condition while limiting identification of variants of uncertain significance and pathogenic variants in genes that do not explain the underlying phenotype. (3) In some laboratories, panel options may include a custom laboratory-designed panel and/or custom phenotype-focused exome analysis that includes genes specified by the clinician. (4) Methods used in a panel may include sequence analysis, deletion/duplication analysis, and/or other non-sequencing-based tests.
Due to the frequency of large deletions or duplications in SLC25A13 associated with this disorder that will not be detected by sequence analysis, a multigene panel that also includes deletion/duplication analysis is recommended (see Table 3).
For an introduction to multigene panels click here. More detailed information for clinicians ordering genetic tests can be found here.
Table 3.
Molecular Genetic Testing Used in Citrin Deficiency
View in own window
| Gene 1 | Method | Proportion of Probands with Pathogenic Variants 2 Detectable by Method |
|---|
|
SLC25A13
| Sequence analysis 3 | 85%-90% 4 |
| Gene-targeted deletion/duplication analysis 5 | 10%-15% 6 |
- 1.
- 2.
- 3.
- 4.
- 5.
Gene-targeted deletion/duplication analysis detects intragenic deletions or duplications. Methods used may include: quantitative PCR, long-range PCR, multiplex ligation-dependent probe amplification (MLPA), and a gene-targeted microarray designed to detect single-exon deletions or duplications.
- 6.
Western Blot Analysis
Western blot analysis for the protein citrin can be considered in the rare instance that neither or only one SLC25A13 pathogenic variant is identified by molecular genetic testing in an individual with biochemical evidence of citrin deficiency.
Western blot analysis using anti-human citrin antibody specific for the amino-terminal half detects little or no cross-reactive immune material in liver,or cultured fibroblasts from individuals with biallelic SLC25A13 pathogenic variants [Takahashi et al 2006, Dimmock et al 2007, Fu et al 2011]. Peripheral blood lymphocytes could be another sample source for western blot analysis [Tokuhara et al 2007], and citrin protein could be detected more easily via western blot using the extracted mitochondria proteins from cultured lymphocytes [Zhang et al 2017].
Clinical Characteristics
Clinical Description
Citrin deficiency can manifest in newborns or infants as neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD), in older children as failure to thrive and dyslipidemia caused by citrin deficiency (FTTDCD), and in adults as recurrent hyperammonemia with neuropsychiatric symptoms in citrullinemia type II (CTLN2). Often FTTDCD and CTLN2 are characterized by the individual's preference for protein-rich and/or lipid-rich foods and aversion to carbohydrate-rich foods. Individuals with CTLN2 may or may not have a prior history of NICCD or FTTDCD. The proportion of persons with NICCD or FTTDCD that evolves into CTLN2 is unknown. For those with NICCD who have no special medical concerns other than dietary management, close medical observation is recommended since they still have biochemical alterations [Song et al 2011] even at the silent stage [Nagasaka et al 2009, Nagasaka et al 2017].
Neonatal Intrahepatic Cholestasis Caused by Citrin Deficiency (NICCD)
Children younger than age one year with NICCD have transient intrahepatic cholestasis (see Table 4). Other findings include diffuse fatty liver with hepatomegaly and parenchymal cellular infiltration associated with hepatic fibrosis, a history of low birth weight, growth restriction, hypoproteinemia, decreased coagulation factors, hemolytic anemia, variable (mainly mild) liver dysfunction, and/or hypoglycemia.
Table 4.
Measurements of Hepatic Cell Function at Age 0-6 Months in NICCD
View in own window
| Assayed Item | Median (25%-75% range)
(mg/dL) | Control Range
(mg/dL) |
|---|
| TB in NICCD | 4.9 (2.8-8.0) | 0.2-1.0 |
| DB in NICCD | 2.5 (1.5-3.7) | 0-0.4 |
| TB/DB ratio in NICCD | 0.55 (0.41-0.66) | — |
| TBA | 239 (172-293) | 5-25 |
| AFP | 91,900 (33,200-174,700) | 260-6,400 1, 2
2-55 2, 3 |
AFP = α-fetoprotein; DB = direct bilirubin; TB = total bilirubin; TBA = total bile acids
- 1.
- 2.
- 3.
NICCD is generally not severe, although liver transplantation has been required in rare instances [Tamamori et al 2002, Kobayashi et al 2006]. Symptoms typically resolve by age one year with treatment, including fat-soluble vitamin supplementation and use of lactose-free therapeutic formulas (for those with secondary galactosemia) and/or medium-chain triglyceride (MCT)-enriched therapeutic formulas [Ohura et al 2003, Song et al 2010, Hayasaka et al 2012, Zhang et al 2014a].
Starting around age one to two years, children show a strong preference for protein-rich and lipid-rich foods and an aversion to sugar-rich and carbohydrate-rich foods [Hachisu et al 2005, Saheki & Kobayashi 2005, Saheki et al 2008].
In the second or later decades, some individuals with citrin deficiency develop severe CTLN2 with neuropsychiatric symptoms [Saheki & Kobayashi 2002]. Typically, the transition from the adaptation (and/or compensation) stage following NICCD to the onset of CTLN2 is gradual, but the CTLN2 presentations usually occur suddenly.
Failure to Thrive and Dyslipidemia Caused by Citrin Deficiency (FTTDCD)
FTTDCD has recently been proposed as a novel post-NICCD phenotype before the onset of CTLN2 [Song et al 2011]. The clinical and laboratory features of FTTDCD are still being established. During this period (traditionally assumed to be an "apparently healthy" stage before CTLN2 onset) some children were found to have laboratory and/or clinical abnormalities.
The laboratory abnormalities include dyslipidemia manifesting as higher levels of triglyceride and total- and LDL-cholesterols, but lower level of HDL-cholesterol [Song et al 2009a, Song et al 2011] and other findings such as increased lactate-to-pyruvate ratio, higher levels of urinary oxidative stress markers, and considerable deviation in tricarboxylic acid (TCA) cycle metabolites [Kobayashi & Saheki 2004, Saheki & Kobayashi 2005, Kobayashi et al 2006, Nagasaka et al 2009, Lee et al 2010, Takeuchi et al 2015, Nagasaka et al 2017].
The clinical abnormalities include growth restriction, hypoglycemia, and pancreatitis. Severe fatigue and impaired quality of life were identified in citrin-deficient children in the adaptation and compensation (traditionally assumed to be "silent") stage [Okano et al 2013]. Moreover, a female age 12 years with citrin deficiency presenting with severe anorexia and body weight loss, mimicking the restricting type of anorexia nervosa, has been reported [Takeuchi et al 2015].
Citrullinemia Type II (CTLN2)
CTLN2 is characterized by recurring episodes of hyperammonemia and neurologic and psychotic symptoms that closely resemble those of hepatic encephalopathy or genetic urea cycle disorders, including nocturnal delirium, aberrant behaviors (aggression, irritability, and hyperactivity), delusions, disorientation, restlessness, drowsiness, loss of memory, flapping tremor, convulsive seizures, and coma. Brain CT is normal, and EEG shows diffuse slow waves.
Presentation is sudden and usually between ages 20 and 50 years (range: 11-79 years; mean age: 34.4 ±12.8 years; n=103) [Yasuda et al 2000].
Many individuals with CTLN2 have a strong preference for protein-rich and/or lipid-rich foods (e.g., beans, peanuts, eggs, milk, cheese, fish, and meat) and an aversion to carbohydrate-rich foods including rice, juice, and sweets. Symptoms are often provoked by alcohol and sugar intake, medication, and/or surgery.
Most individuals are thin. More than 90% have a body mass index lower than 20 and approximately 40% have a body mass index lower than 17 (range: 15.6-19.1; n=110) [Kobayashi et al 2006] (range in healthy Japanese individuals: 20-24 in males; 19-23 in females).
The following complications occur in more than 10% of individuals with CTLN2 [Kobayashi et al 2000].
Pancreatitis. Juvenile-onset chronic pancreatitis and hepatocellular carcinoma without cirrhosis can precede the appearance of CTLN2 [
Ikeda et al 2004].
Hyperlipidemia. Hypertriglyceridemia is frequently observed if high-carbohydrate meals are provided to individuals with citrin deficiency [
Imamura et al 2003].
Intrahepatic cholestasis is rare; however, some individuals are noted in retrospect to have had signs of NICCD in early childhood [Kobayashi & Saheki 2004, Saheki & Kobayashi 2005]. For example, a 16-year-old with CTLN2 undergoing liver transplantation [Kasahara et al 2001] had had transient hypoproteinemia and jaundice in early infancy [Tomomasa et al 2001].
Abnormal laboratory findings
Pathologic findings include fatty infiltration and mild fibrosis of the liver despite little or no liver dysfunction.
Penetrance
There appears to be a difference in penetrance of the CTLN2 phenotype related to the sex of the individual.
The male-to-female ratio in NICCD is roughly equal (73:80) while the male-to-female ratio in CTLN2 is 2.4 to 1 (120:50) [
Kobayashi & Saheki 2004].
The unequal male-to-female ratio in CTLN2 suggests that for unknown reasons, among individuals with
biallelic SLC25A13 pathogenic variants, females are more resistant to the CTLN2
phenotype than males.
Nomenclature
NICCD. NICCD was known as "idiopathic neonatal hepatitis with fatty liver of unknown origin" [Ohura et al 1997] before molecular genetic testing confirmed the presence of biallelic SLC25A13 pathogenic variants.
CTLN2.
Miyakoshi et al [1968] reported that blood citrulline concentrations were increased in individuals with hyperammonemia and a unique chronic recurrent hepatocerebral degeneration. This hepatocerebral degeneration came to be known as "pseudo-ulegyric hepatocerebral disease" on the basis of pathologic brain changes, and "nutritional hepatocerebral disease" on the basis of metabolic disturbance resulting from a highly unbalanced diet or developmental disturbance caused by endocrine abnormalities.
Saheki et al [1981] reported this hepatocerebral disease as a type of citrullinemia with a qualitative and liver-specific decrease of the argininosuccinate synthetase activity/protein, and later Saheki et al [1985] named it "adult-onset type II citrullinemia."
Prevalence
In Japan, the frequency of homozygotes or compound heterozygotes for SLC25A13 pathogenic variants is calculated at 1:17,000 based on the carrier (heterozygote) rate of 1:65 [Saheki & Kobayashi 2002, Tabata et al 2008]. This is similar to the observed prevalence of NICCD [Shigematsu et al 2002], but different from the observed prevalence of CTLN2 (1:100,000-1:230,000) [Kobayashi et al 2006]. Based on their observations, the authors believe that most individuals of Japanese heritage with biallelic SLC25A13 pathogenic variants have NICCD.
Until recently, citrin deficiency was thought to be restricted to Japan; citrin deficiency is now recognized to be pan ethnic [Dimmock et al 2009]. Individuals with novel SLC25A13 pathogenic variants have been identified in Israel, Pakistan, the United States, the United Kingdom, China, and the Czech Republic [Ben-Shalom et al 2002, Hutchin et al 2006, Luder et al 2006, Dimmock et al 2007, Fiermonte et al 2008, Song et al 2008, Tabata et al 2008, Song et al 2009b, Song et al 2011].
The carrier frequency is also high in China (1:65), especially southern China including Taiwan (1:48), and in Korea (1:112) [Lu et al 2005, Lee et al 2011].
Differential Diagnosis
Plasma concentration of citrulline, increased in citrin deficiency, is also increased in the following disorders:
Hyperammonemia, seen in citrin deficiency, also occurs in the urea cycle disorders, which result from defects in the metabolism of the nitrogen produced by the breakdown of protein and other nitrogen-containing molecules (see Urea Cycle Disorders Overview). Severe deficiency or total absence of activity of any of the first four enzymes (CPSI, OTC, ASS, ASL) in the urea cycle, the ornithine transporter, or the cofactor producer (NAGS) results in the accumulation of ammonia and other precursor metabolites during the first few days of life in most affected individuals.
Neonatal/infantile cholestasis, seen in citrin deficiency, also occurs in the following disorders:
Idiopathic neonatal hepatitis (INH) and extrahepatic biliary atresia (EBA). In comparison with INH and EBA, NICCD is associated with lower levels of serum direct bilirubin or ALT and higher levels of serum total bile acids and alkaline phosphatase. NICCD also has higher levels of serum γ-GTP and lower levels of serum AST activity than are seen in INH [
Tazawa et al 2005].
Progressive familial intrahepatic cholestasis (PFIC). The high-serum γ-GTP levels of NICCD may distinguish it from other intrahepatic cholestasis disorders with low-normal γ-GTP levels including PFIC and benign recurrent intrahepatic cholestasis (BRIC). PFIC1 is caused by
biallelic pathogenic variants in
ATP8B1 (see
ATP8B1 Deficiency); PFIC2 is caused by biallelic pathogenic variants in
ABCB11 (OMIM
601847). Some cases of BRIC are caused by biallelic pathogenic variants in
ATP8B1.
Hereditary jaundice and hyperbilirubinemia result from defects in the metabolism of bilirubin. These include disorders resulting in predominantly unconjugated (indirect) hyperbilirubinemia (UDP-glucuronosyltransferase 1-1 deficiency) and those resulting in predominantly conjugated (direct) hyperbilirubinemia (deficiency in canalicular ATP-dependent transporters: ABCC2 [OMIM 601107], ABCB11 [OMIM 603201], or ATP8B1 [see ATP8B1 Deficiency]).
Other
Portal-systemic shunts can be excluded by angiography. These shunts refer to abnormal blood flow from the portal to the postcaval, hepatic, or splenic vein. Patients with citrin deficiency have no such shunts on medical imaging tests (e.g., sonography, MRI).
More than 30% of individuals with CTLN2 have been misdiagnosed initially as having epileptic seizures and/or a psychological disorder (e.g., depression, schizophrenia); others may be diagnosed as having diseases such as hepatoma, pancreatitis, and hyperlipidemia.
Management
Evaluations Following Initial Diagnosis
To establish the extent of disease and needs of an individual diagnosed with a phenotype of citrin deficiency, the following are recommended:
NICCD
Assess the size of the liver and spleen.
Seek evidence of fatty liver by abdominal US, CT, or MRI.
Investigate feeding pattern.
FTTDCD
CTLN2. Investigate carbohydrate, protein, and lipid composition of the diet.
All phenotypes. Consult with a clinical geneticist and/or genetic counselor.
Treatment of Manifestations
NICCD. The symptoms in most children with NICCD resolve by age 12 months following supplementation with fat-soluble vitamins and use of lactose-free and MCT-enriched therapeutic formulas [Ohura et al 2003, Song et al 2010, Hayasaka et al 2012, Zhang et al 2014a].
Two sibs improved after switching from breast milk to formula, which has higher proline content [Ben-Shalom et al 2002].
Some children with NICCD improve without treatment, which could be the effect of reduction of breast milk and/or common formulas while simultaneously introducing solid supplements such as eggs and meat, which are rich in protein and lipid and therefore beneficial for citrin-deficient individuals [Song et al 2010].
The treatment with therapeutic formulas is not lifelong. Most infants with NICCD will recover clinically and biochemically by age one year, at which point protein- and lipid-enriched textured or solid supplements could be introduced. Whether continued treatment beyond a year can reduce the likelihood of the FTTDCD and CTLN2 phenotype is currently unknown.
Moreover, zinc deficiency is common in NICCD, and thus zinc supplementation should be encouraged when laboratory evidence indicates zinc deficiency, especially in individuals with marked failure to thrive.
Four infants with NICCD and severe liver dysfunction were diagnosed as having tyrosinemia of unknown cause and underwent liver transplantation at age ten to 12 months [Tamamori et al 2002, Kobayashi et al 2006].
FTTDCD. Few treatment measures have been described for this novel citrin-deficient phenotype.
A toddler with FTTDCD was fed in accordance with his own food preferences (including aversion to rice and fondness for fish); FTT improved gradually, with weight for age recovering beyond the third percentile at age three years. The dyslipidemia also improved gradually [
Song et al 2009a].
In addition to dietary treatment, administration of sodium pyruvate may be effective in correcting growth restriction [
Mutoh et al 2008,
Saheki et al 2010]. Sodium pyruvate reduces the NADH/NAD
+ ratio in the hepatocyte, a pivotal alteration for citrin deficiency development, and this could be associated with its ameliorating effect on growth restriction.
CTLN2. The most successful therapy to date has been liver transplantation [Ikeda et al 2001, Kasahara et al 2001, Yazaki et al 2004, Hirai et al 2008], which prevents episodic hyperammonemic crises, corrects the metabolic disturbances, and eliminates preference for protein-rich foods [Kobayashi & Saheki 2004]. Nearly all individuals with CTLN2 required liver transplantation in the past; however, the introduction of arginine and sodium pyruvate and medium chain triglyceride (MCT) oil administration has altered the situation. Other treatments include the following:
Prevention of Secondary Complications
Vitamin D deficiency and zinc deficiency are common complications in NICCD [Song et al, personal communication]. Severe infection and liver cirrhosis have also been reported to be lethal complications in some individuals with NICCD. Therefore, vitamin D and zinc supplements and active infection control are recommended in persons with NICCD.
Surveillance
To monitor for emergence of the FTTDCD phenotype in persons with citrin deficiency older than age one year, close surveillance of anthropometric indices (e.g., height, weight, and head circumference; serum lipid levels including triglycerides, total cholesterol, HDL-cholesterol, and LDL-cholesterol) is appropriate.
It is recommended that the following be measured every several months:
Increases in plasma citrulline concentration and serum PSTI suggest onset of CTLN2 [Tsuboi et al 2001, Mutoh et al 2008] and should prompt initiation of treatment.
Agents/Circumstances to Avoid
Low-protein/high-caloric (high-carbohydrate) diet. Although a low-protein/high-caloric diet helps prevent hyperammonemia in urea cycle enzyme deficiencies, it is harmful for individuals with all forms of citrin deficiency (i.e., NICCD, FTTDCD, or CTLN2) [Saheki et al 2004, Saheki & Kobayashi 2005, Saheki et al 2006]. A high-carbohydrate diet may increase NADH production, disturb urea synthesis, and stimulate the citrate-malate shuttle, resulting in hyperammonemia, fatty liver, and hypertriglyceridemia [Saheki & Kobayashi 2002, Imamura et al 2003, Saheki et al 2006, Saheki et al 2007].
Infusion of sugars including glycerol, fructose, and glucose. Severe brain edema treated with glycerol-containing osmotic agents has resulted in continued deterioration and is contraindicated in those with CTLN2 [Yazaki et al 2005]. Degradation of large amounts of glycerol and fructose generates NADH in liver cytosol, which may disturb liver function [Saheki et al 2004, Yazaki et al 2005, Takahashi et al 2006].
Infusion of high-concentration glucose may also exacerbate hyperammonemia [Tamakawa et al 1994, Takahashi et al 2006].
Note: Mannitol infusion appears to be safer [Yazaki et al 2005].
Alcohol. Drinking alcohol can trigger the onset of CTLN2 because alcohol dehydrogenase (ADH) generates NADH in the cytosol of the liver.
Medications. Acetaminophen and rabeprozole may trigger CTLN2 [Shiohama et al 1993, Imamura et al 2003].
Evaluation of Relatives at Risk
It is appropriate to clarify the genetic status of at-risk asymptomatic sibs of a proband for citrin deficiency so that appropriate dietary management of infants (discontinuation of breast feeding and introduction of lactose-free and/or MCT-enriched formulas) can be instituted before symptoms occur.
Because asymptomatic or presymptomatic individuals with citrin deficiency do not always show biochemical abnormalities, definitive diagnosis of at-risk relatives (e.g., sibs) would depend heavily on SLC25A13 molecular genetic findings in the proband:
If only one
SLC25A13 pathogenic variant is ascertained in the index case, it may not be feasible to definitively exclude the diagnosis in at-risk relatives who have the one identified pathogenic variant. In such cases, serial clinical and biochemical assessment will be needed over time to confirm or rule out the diagnosis.
If no
SLC25A13 pathogenic variant is ascertained in the index case, molecular testing of at-risk individuals will not be helpful. In such cases, serial clinical and biochemical assessment will be needed over time to confirm or rule out the diagnosis
See Genetic Counseling for issues related to testing of at-risk relatives for genetic counseling purposes.
Therapies Under Investigation
Search ClinicalTrials.gov in the US and EU Clinical Trials Register in Europe for access to information on clinical studies for a wide range of diseases and conditions. Note: There may not be clinical trials for this disorder.
Genetic Counseling
Genetic counseling is the process of providing individuals and families with
information on the nature, mode(s) of inheritance, and implications of genetic disorders to help them
make informed medical and personal decisions. The following section deals with genetic
risk assessment and the use of family history and genetic testing to clarify genetic
status for family members; it is not meant to address all personal, cultural, or
ethical issues that may arise or to substitute for consultation with a genetics
professional. —ED.
Risk to Family Members
Parents of a proband
Sibs of a proband
Offspring of a proband. Unless an individual with citrin deficiency has children with an affected individual or a carrier, his/her offspring will be obligate heterozygotes (carriers) for an SLC25A13 pathogenic variant.
Other family members. Each sib of the proband's parents is at a 50% risk of being a carrier of an SLC25A13 pathogenic variant.
Carrier Detection
Carrier testing for at-risk relatives requires prior identification of the SLC25A13 pathogenic variants in the family.
Prenatal Testing and Preimplantation Genetic Testing
Once the SLC25A13 pathogenic variants have been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic testing are possible.
Differences in perspective may exist among medical professionals and within families regarding the use of prenatal testing. While most centers would consider use of prenatal testing to be a personal decision, discussion of these issues may be helpful.
Resources
GeneReviews staff has selected the following disease-specific and/or umbrella
support organizations and/or registries for the benefit of individuals with this disorder
and their families. GeneReviews is not responsible for the information provided by other
organizations. For information on selection criteria, click here.
MedlinePlus
Children's Liver Disease Foundation
United Kingdom
Phone: +44 (0) 121 212 3839
Email: info@childliverdisease.org
Metabolic Support UK
United Kingdom
Phone: 0845 241 2173
Molecular Genetics
Information in the Molecular Genetics and OMIM tables may differ from that elsewhere in the GeneReview: tables may contain more recent information. —ED.
Table A.
Citrin Deficiency: Genes and Databases
View in own window
Data are compiled from the following standard references: gene from
HGNC;
chromosome locus from
OMIM;
protein from UniProt.
For a description of databases (Locus Specific, HGMD, ClinVar) to which links are provided, click
here.
Table B.
View in own window
|
603471 | CITRULLINEMIA, TYPE II, ADULT-ONSET; CTLN2 |
|
603859 | SOLUTE CARRIER FAMILY 25 (CITRIN), MEMBER 13; SLC25A13 |
|
605814 | CITRULLINEMIA, TYPE II, NEONATAL-ONSET |
Gene structure.
SLC25A13 comprises 18 exons [Kobayashi et al 1999, Sinasac et al 1999]. For a detailed summary of gene and protein information, see Table A, Gene.
Pathogenic variants. To date, more than 100 pathogenic variants occurring in exons or introns resulting in pathogenic missense variants, predicted truncated forms of citrin, or abnormal mRNA splicing have been reported [Kobayashi et al 1999, Lin et al 2012, Takahashi et al 2012, Chen et al 2013, Song et al 2013, Tong et al 2013, Wen et al 2013, Zeng et al 2014, Zhang et al 2014a, Zhang et al 2014b, Bijarnia-Mahay et al 2015, Wang et al 2015, Xiong et al 2015, Lin et al 2016, Zeng et al 2016, Zheng et al 2016, Oh et al 2017, Zhang et al 2017].
Some of the 20 pathogenic variants identified in Japanese individuals have been found in Chinese, Vietnamese, and Korean individuals with citrin deficiency (NICCD or CTLN2) [Lu et al 2005, Lee et al 2006, Song et al 2006, Tsai et al 2006, Yeh et al 2006, Ko et al 2007a, Ko et al 2007b, Song et al 2008, Tabata et al 2008].
Different pathogenic variants were found in Israel, the United States, the United Kingdom, and China [Ben-Shalom et al 2002, Hutchin et al 2006, Luder et al 2006, Dimmock et al 2007, Song et al 2008, Tabata et al 2008, Song et al 2009b, Xing et al 2010, Fu et al 2011, Song et al 2011].
Table 5.
Selected SLC25A13 Pathogenic Variants
View in own window
Variants listed in the table have been provided by the authors. GeneReviews staff have not independently verified the classification of variants.
GeneReviews follows the standard naming conventions of the Human Genome Variation Society (varnomen.hgvs.org). See Quick Reference for an explanation of nomenclature.
- 1.
Variant designation that does not conform to current naming conventions
- 2.
Normal gene product. Citrin and its homolog aralar [del Arco & Satrústegui 1998] are members of the SLC25 (solute carrier family 25) protein family. Both proteins are localized in the mitochondrial inner membrane and function as a Ca2+-binding/-stimulated aspartate-glutamate carrier (AGC), a component of the malate-aspartate NADH shuttle [Palmieri et al 2001, Kobayashi & Saheki 2003].
Citrin is expressed in the liver; aralar in the brain and skeletal muscle; both are expressed in the kidney and heart [Kobayashi et al 1999]. Citrin as a liver-type AGC plays a role in various metabolic pathways, including aerobic glycolysis, gluconeogenesis, the urea cycle, and protein and nucleotide syntheses [Saheki & Kobayashi 2002, Saheki et al 2004, Saheki & Kobayashi 2005, Saheki et al 2006].
Abnormal gene product. Most SLC25A13 pathogenic variants cause or predict truncation of the citrin protein or delete a loop between the mitochondrial transmembrane domains. The lack of significant citrin protein was confirmed by western blot analysis using antibody against the N-terminal half of the human citrin protein, which detected little or no cross-reactive immune material in liver, cultured fibroblasts, and lymphocytes from individuals with biallelic SLC25A13 pathogenic variants [Yasuda et al 2000, Takahashi et al 2006, Dimmock et al 2007, Tokuhara et al 2007, Fu et al 2011].
References
Literature Cited
Ben-Shalom E, Kobayashi K, Shaag A, Yasuda T, Gao HZ, Saheki T, Bachmann C, Elpeleg O. Infantile citrullinemia caused by citrin deficiency with increased dibasic amino acids.
Mol Genet Metab. 2002;77:202–8. [
PubMed: 12409267]
Bijarnia-Mahay S, Häberle J, R, Fenacht VR, Shigematsu Y, Saxena R, Verma IC. Citrin deficiency: A treatable cause of acute psychosis in adults.
Neurol India. 2015;63:220–2. [
PubMed: 25947987]
Chen R, Wang XH, Fu HY, Zhang SR, Abudouxikuer K, Saheki T, Wang JS. Different regional distribution of SLC25A13 mutations in Chinese patients with neonatal intrahepatic cholestasis.
World J Gastroenterol. 2013;19:4545–51. [
PMC free article: PMC3725380] [
PubMed: 23901231]
del Arco A, Satrústegui J. Molecular cloning of Aralar, a new member of the mitochondrial carrier superfamily that binds calcium and is present in human muscle and brain.
J Biol Chem. 1998;273:23327–34. [
PubMed: 9722566]
Dimmock D, Kobayashi K, Iijima M, Tabata A, Wong LJ, Saheki T, Lee B, Scaglia F. Citrin deficiency: a novel cause of failure to thrive that responds to a high protein, low carbohydrate diet.
Pediatrics. 2007;119:e773–7. [
PubMed: 17332192]
Dimmock D, Maranda B, Dionisi-Vici C, Wang J, Kleppe S, Fiermonte G, Bai R, Hainline B, Hamosh A, O'Brien WE, Scaglia F, Wong LJ. Citrin deficiency, a perplexing global disorder.
Mol Genet Metab. 2009;96:44–9. [
PubMed: 19036621]
Feillet F, Merten M, Battaglia-Hsu SF, Rabier D, Kobayashi K, Straczek J, Brivet M, Favre E, Guéant JL. Evidence of cataplerosis in a patient with neonatal classical galactosemia presenting as citrin deficiency.
J Hepatol. 2008;48:517–22. [
PubMed: 18207281]
Fiermonte G, Soon D, Chaudhuri A, Paradies E, Lee PJ, Krywawych S, Palmieri F, Lachmann RH. An adult with type 2 citrullinemia presenting in Europe.
N Engl J Med. 2008;358:1408–9. [
PubMed: 18367750]
Fu HY, Zhang SR, Wang XH, Saheki T, Kobayashi K, Wang JS. The mutation spectrum of the SLC25A13 gene in Chinese infants with intrahepatic cholestasis and aminoacidemia.
J Gastroenterol. 2011;46:510–8. [
PubMed: 20927635]
Fukumoto K, Sumida Y, Yoshida N, Sakai K, Kanemasa K, Itoh Y, Mitsufuji S, Kataoka K, Okanoue T. A case of adult-onset type II citrullinemia having a liver histology of nonalcoholic steatohepatitis (NASH).
Nippon Shokakibyo Gakkai Zasshi. 2008;105:244–51. [
PubMed: 18250596]
Gao HZ, Kobayashi K, Tabata A, Tsuge H, Iijima M, Yasuda T, Kalkanoglu HS, Dursun A, Tokatli A, Coskun T, Trefz FK, Skladal D, Mandel H, Seidel J, Kodama S, Shirane S, Ichida T, Makino S, Yoshino M, Kang JH, Mizuguchi M, Barshop BA, Fuchinoue S, Seneca S, Zeesman S, Knerr I, Rodes M, Wasant P, Yoshida I, De Meirleir L, Abdul Jalil M, Begum L, Horiuchi M, Katunuma N, Nakagawa S, Saheki T. Identification of 16 novel mutations in the argininosuccinate synthetase gene and genotype-phenotype correlation in 38 classical citrullinemia patients.
Hum Mutat. 2003;22:24–34. [
PubMed: 12815590]
Hachisu M, Oda Y, Goto M, Kobayashi K, Saheki T, Ohura T, Noma S, Kitanaka S. Citrin deficiency presenting with ketotic hypoglycaemia and hepatomegaly in childhood.
Eur J Pediatr. 2005;164:109–10. [
PubMed: 15592876]
Hagiwara N, Sekijima Y, Takei Y, Ikeda S, Kawasaki S, Kobayashi K, Saheki T. Hepatocellular carcinoma in a case of adult-onset type II citrullinemia.
Intern Med. 2003;42:978–82. [
PubMed: 14606711]
Hayasaka K, Numakura C, Toyota K, Kakizaki S, Watanabe H, Haga H, Takahashi H, Takahashi Y, Kaneko M, Yamakawa M, Nunoi H, Kato T, Ueno Y, Mori M. Medium-chain triglyceride supplementation under a low-carbohydrate formula is a promising therapy for adult-onset type II citrullinemia.
Mol Genet Metab Rep. 2014;1:42–50. [
PMC free article: PMC5121258] [
PubMed: 27896073]
Hayasaka K, Numakura C, Toyota K, Kimura T. Treatment with lactose (galactose)-restricted and medium-chain triglyceride-supplemented formula for neonatal intrahepatic cholestasis caused by citrin deficiency.
JIMD Rep. 2012;2:37–44. [
PMC free article: PMC3509838] [
PubMed: 23430852]
Hirai I, Kimura W, Suto K, Fzjimoto H, Watanabe T, Fuse A, Kobayashi K, Iijima M, Saheki T, Nakatsuka T, Sugawara Y, Makuuchi M. Living donor liver transplantation for type II citrullinemia from a heterozygous donor.
Hepatogastroenterology. 2008;55:2211–6. [
PubMed: 19260507]
Hutchin T, Preece MA, Kobayashi K, Saheki T, Brown R, Kelly DA, McKiernan PJ, Green A, Baumann U. Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) in a European patient. J Inherit Metab Dis. 2006;29 Suppl 1:112.
Ikeda S, Kawa S, Takei Y, Yamamoto K, Shimojo H, Tabata K, Kobayashi K, Saheki T. Chronic pancreatitis associated with adult-onset type II citrullinemia: clinical and pathologic findings.
Ann Intern Med. 2004;141:W109-10. [
PubMed: 15466765]
Ikeda S, Yazaki M, Takei Y, Ikegami T, Hashikura Y, Kawasaki S, Iwai M, Kobayashi K, Saheki T. Type II (adult onset) citrullinaemia: clinical pictures and the therapeutic effect of liver transplantation.
J Neurol Neurosurg Psychiatry. 2001;71:663–70. [
PMC free article: PMC1737600] [
PubMed: 11606680]
Imamura Y, Kobayashi K, Shibatou T, Aburada S, Tahara K, Kubozono O, Saheki T. Effectiveness of carbohydrate-restricted diet and arginine granules therapy for adult-onset type II citrullinemia: a case report of siblings showing homozygous SLC25A13 mutation with and without the disease.
Hepatol Res. 2003;26:68–72. [
PubMed: 12787807]
Kasahara M, Ohwada S, Takeichi T, Kaneko H, Tomomasa T, Morikawa A, Yonemura K, Asonuma K, Tanaka K, Kobayashi K, Saheki T, Takeyoshi I, Morishita Y. Living-related liver transplantation for type II citrullinemia using a graft from heterozygote donor.
Transplantation. 2001;71:157–9. [
PubMed: 11211185]
Ko JM, Kim GH, Kim JH, Kim JY, Choi JH, Ushikai M, Saheki T, Kobayashi K, Yoo HW. Six cases of citrin deficiency in Korea.
Int J Mol Med. 2007a;20:809–15. [
PubMed: 17982687]
Kobayashi K, Horiuchi M, Saheki T. Pancreatic secretory trypsin inhibitor as a diagnostic marker for adult-onset type II citrullinemia.
Hepatology. 1997;25:1160–5. [
PubMed: 9141434]
Kobayashi K, Iijima M, Ushikai M, Ikeda S, Saheki T. Citrin deficiency. J Jpn Pediatr Soc. 2006;110:1047–59.
Kobayashi K, Iijima M, Yasuda T, Sinasac DS, Yamaguchi N, Tsui L-C, Scherer SW, Saheki T. Type II citrullinemia (citrin deficiency): a mysterious disease caused by a defect of calcium-binding mitochondrial carrier protein. In: Pochet R, ed. Calcium: The Molecular Basis of Calcium Action in Biology and Medicine. Dordrecht, Netherlands: Kluwer; 2000:565-87.
Kobayashi K, Saheki T. Molecular basis of citrin deficiency.
Seikagaku. 2004;76:1543–59. [
PubMed: 15675368]
Kobayashi K, Saheki T. Aspartate glutamate carrier (citrin) deficiency. In: Broer S, Wagner CS, eds. Membrane Transporter Diseases. New York, NY: Kluwer Academic/Plenum Publishers; 2003:147-60.
Kobayashi K, Sinasac DS, Iijima M, Boright AP, Begum L, Lee JR, Yasuda T, Ikeda S, Hirano R, Terazono H, Crackower MA, Kondo I, Tsui LC, Scherer SW, Saheki T. The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein.
Nat Genet. 1999;22:159–63. [
PubMed: 10369257]
Komatsu M, Yazaki M, Tanaka N, Sano K, Hashimoto E, Takei Y, Song YZ, Tanaka E, Kiyosawa K, Saheki T, Aoyama T, Kobayashi K. Citrin deficiency as a cause of chronic liver disorder mimicking non-alcoholic fatty liver disease.
J Hepatol. 2008;49:810–20. [
PubMed: 18620775]
Lee BH, Jin HY, Kim GH, Choi JH, Yoo HW. Nonalcoholic fatty liver disease in 2 siblings with adult-onset type II citrullinemia.
J Pediatr Gastroenterol Nutr. 2010;50:682–5. [
PubMed: 20400906]
Lee HC, Mak CM, Lam CW, Yuen YP, Chan AO, Shek CC, Siu TS, Lai CK, Ching CK, Siu WK, Chen SP, Law CY, Tai HL, Tam S, Chan AY. Analysis of inborn errors of metabolism: disease spectrum for expanded newborn screening in Hong Kong.
Chin Med J (Engl). 2011;124:983–9. [
PubMed: 21542954]
Lee NC, Chien YH, Kobayashi K, Saheki T, Chen H-L, Chiu PC, Ni YH, Chang MH, Hwu WL. Time course of acylcarnitine elevation in neonatal intrahepatic cholestasis caused by citrin deficiency.
J Inherit Metab Dis. 2006;29:551–5. [
PubMed: 16736097]
Lin WX, Zeng HS, Zhang ZH, Mao M, Zheng QQ, Zhao ST, Cheng Y, Chen FP, Wen WR, Song YZ. Molecular diagnosis of pediatric patients with citrin deficiency in China: SLC25A13 mutation spectrum and the geographic distribution.
Sci Rep. 2016;6:29732. [
PMC free article: PMC4942605] [
PubMed: 27405544]
Lin WX, Zhang ZH, Deng M, Cai XR, Song YZ. Multiple ovarian antral follicles in a preterm infant with neonatal intrahepatic cholestasis caused by citrin deficiency: A clinical, genetic and transcriptional analysis.
Gene. 2012;505:269–75. [
PubMed: 22710133]
Lu YB, Kobayashi K, Ushikai M, Tabata A, Iijima M, Li MX, Lei L, Kawabe K, Taura S, Yang Y, Liu TT, Chiang SH, Hsiao KJ, Lau YL, Tsui LC, Lee DH, Saheki T. Frequency and distribution in East Asia of 12 mutations identified in the SLC25A13 gene of Japanese patients with citrin deficiency.
J Hum Genet. 2005;50:338–46. [
PubMed: 16059747]
Luder AS, Tabata A, Iijima M, Kobayashi K, Mandel H. Citrullinaemia type 2 outside East Asia: Israeli experience. J Inherit Metab Dis. 2006;29:59.
Miyakoshi T, Takahashi T, Kato M, Watanabe M, Ito C. Abnormal citrulline metabolism of Inose-type hepatocerebral disease. Shinkei Kagaku. 1968;7:88–91.
Mutoh K, Kurokawa K, Kobayashi K, Saheki T. Treatment of a citrin-deficient patient at the early stage of adult-onset type II citrullinaemia with arginine and sodium pyruvate.
J Inherit Metab Dis. 2008;31 Suppl 2:S343–7. [
PubMed: 18958581]
Nagasaka H, Komatsu H, Inui A, Nakacho M, Morioka I, Tsukahara H, Kaji S, Hirayama S, Miida T, Kondou H, Ihara K, Yagi M, Kizaki Z, Bessho K, Kodama T, Iijima K, Saheki T, Yorifuji T, Honda A. Circulating tricarboxylic acid cycle metabolite levels in citrin-deficient children with metabolic adaptation, with and without sodium pyruvate treatment.
Mol Genet Metab. 2017;120:207–12. [
PubMed: 28041819]
Nagasaka H, Okano Y, Tsukahara H, Shigematsu Y, Momoi T, Yorifuji J, Miida T, Ohura T, Kobayashi K, Saheki T, Hirano K, Takayanagi M, Yorifuji T. Sustaining hypercitrullinemia, hypercholesterolemia and augmented oxidative stress in Japanese children with aspartate/glutamate carrier isoform 2-citrin-deficiency even during the silent period.
Mol Genet Metab. 2009;97:21–26. [
PubMed: 19232506]
Oh SH, Lee BH, Kim GH, Choi JH, Kim KM, Yoo HW. Biochemical and molecular characteristics of citrin deficiency in Korean children.
J Hum Genet. 2017;62:305–7. [
PubMed: 27829683]
Ohura T, Abukawa D, Aikawa J, Nakagawa M, Ishizawa S, Tazawa Y, Narisawa K, Iinuma K. Disturbances of amino acid metabolism in infants with idiopathic neonatal hepatitis. J Jpn Pediatr Soc. 1997;101:1522–5.
Ohura T, Kobayashi K, Abukawa D, Tazawa Y, Aikawa J, Sakamoto O, Saheki T, Iinuma K. A novel inborn error of metabolism detected by elevated methionine and/or galactose in newborn screening: neonatal intrahepatic cholestasis caused by citrin deficiency.
Eur J Pediatr. 2003;162:317–22. [
PubMed: 12692712]
Ohura T, Kobayashi K, Tazawa Y, Abkawa D, Sakamoto O, Tsuchiya S, Saheki T. Clinical pictures of 75 patients with neonatal intrahepatic cholestasis caused by citrin deficiency.
J Inherit Metab Dis. 2007;30:139–44. [
PubMed: 17323144]
Okano Y, Kobayashi K, Ihara K, Ito T, Yoshino M, Watanabe Y, Kaji S, Ohura T, Nagao M, Noguchi A, Mushiake S, Hohashi N, Hashimoto-Tamaoki T. Fatigue and quality of life in citrin deficiency during adaptation and compensation stage.
Mol Genet Metab. 2013;109:9–13. [
PubMed: 23453692]
Palmieri L, Pardo B, Lasorsa FM, del Arco A, Kobayashi K, Iijima M, Runswick MJ, Walker JE, Saheki T, Satrustegui J, Palmieri F. Citrin and aralar1 are Ca(2+)-stimulated aspartate/glutamate transporters in mitochondria.
EMBO J. 2001;20:5060–9. [
PMC free article: PMC125626] [
PubMed: 11566871]
Saheki T, Iijima M, Li MX, Kobayashi K, Horiuchi M, Ushikai M, Okumura F, Meng XJ, Inoue I, Tajima A, Moriyama M, Eto K, Kadowaki T, Sinasac DS, Tsui L-C, Tsuji M, Okano A, Kobayashi T. Citrin/mitochondrial glycerol-3-phosphate dehydrogenase double-knockout mice recapitulate features of human citrin deficiency.
J Biol Chem. 2007;282:25041–52. [
PubMed: 17591776]
Saheki T, Inoue K, Tushima A, Mutoh K, Kobayashi K. Citrin deficiency and current treatment concepts.
Mol Genet Metab. 2010;100:S59–64. [
PubMed: 20233664]
Saheki T, Kobayashi K. Mitochondrial aspartate glutamate carrier (citrin) deficiency as the cause of adult-onset type II citrullinemia (CTLN2) and idiopathic neonatal hepatitis (NICCD).
J Hum Genet. 2002;47:333–41. [
PubMed: 12111366]
Saheki T, Kobayashi K. Physiological role of citrin, a liver-type mitochondrial aspartate-glutamate carrier, and pathophysiology of citrin deficiency. Recent Res Devel Life Sci. 2005;3:59–73.
Saheki T, Kobayashi K, Iijima M, Horiuchi M, Begum L, Jalil MA, Li MX, Lu YB, Ushikai M, Tabata A, Moriyama M, Hsiao KJ, Yang Y. Adult-onset type II citrullinemia and idiopathic neonatal hepatitis caused by citrin deficiency: involvement of the aspartate glutamate carrier for urea synthesis and maintenance of the urea cycle.
Mol Genet Metab. 2004;81 Suppl 1:S20–6. [
PubMed: 15050970]
Saheki T, Kobayashi K, Iijima M, Li MX, Horiuchi M, Tabata A, Lu YB, Ushikai M, Terashi M, Moriyama M. Pathophysiology of citrin deficiency. In: Haussinger D, Kircheis G, Schliess F, eds. Hepatic Encephalopathy and Nitrogen Metabolism. Dordrecht, Netherlands: Springer; 2006:320-8.
Saheki T, Kobayashi K, Terashi M, Ohura T, Yanagawa Y, Okano Y, Hattori T, Fujimoto H, Mutoh K, Kizaki Z, Inui A. Reduced carbohydrate intake in citrin-deficient subjects.
J Inherit Metab Dis. 2008;31:386–94. [
PubMed: 18415701]
Saheki T, Nakano K, Kobayashi K, Imamura Y, Itakura Y, Sase M, Hagihara S, Matuo S. Analysis of the enzyme abnormality in eight cases of neonatal and infantile citrullinaemia in Japan.
J Inherit Metab Dis. 1985;8:155–6. [
PubMed: 3939592]
Saheki T, Ueda A, Hosoya M, Kusumi K, Takada S, Tsuda M, Katsunuma T. Qualitative and quantitative abnormalities of argininosuccinate synthetase in citrullinemia.
Clin Chim Acta. 1981;109:325–35. [
PubMed: 6784969]
Shigematsu Y, Hirano S, Hata I, Tanaka Y, Sudo M, Sakura N, Tajima T, Yamaguchi S. Newborn mass screening and selective screening using electrospray tandem mass spectrometry in Japan.
J Chromatogr B Analyt Technol Biomed Life Sci. 2002;776:39–48. [
PubMed: 12127323]
Shiohama N, Sugita Y, Imamura N, Sato T, Mizuno Y. Type II citrullinemia triggered by acetaminophen.
No To Shinkei. 1993;45:865–70. [
PubMed: 8217412]
Sinasac DS, Crackower MA, Lee JR, Kobayashi K, Saheki T, Scherer SW, Tsui LC. Genomic structure of the adult-onset type II citrullinemia gene, SLC25A13, and cloning and expression of its mouse homologue.
Genomics. 1999;62:289–92. [
PubMed: 10610724]
Soeda J, Yazaki M, Nakata T, Miwa S, Ikeda S, Hosoda W, Iijima M, Kobayashi K, Saheki T, Kojiro M, Miyagawa S. Primary liver carcinoma exhibiting dual hepatocellular-biliary epithelial differentiations associated with citrin deficiency: a case report.
J Clin Gastroenterol. 2008;42:855–60. [
PubMed: 18385606]
Song YZ, Deng M, Chen FP, Wen F, Guo L, Cao SL, Gong J, Xu H, Jiang GY, Zhong L, Kobayashi K, Saheki T, Wang ZN. Genotypic and phenotypic features of citrin deficiency: Five-year experience in a Chinese pediatric center.
Int J Mol Med. 2011;28:33–40. [
PubMed: 21424115]
Song YZ, Guo L, Yang YL, Han LS, Kobayashi K, Saheki T. Failure to thrive and dyslipidemia caused by citrin deficiency: a novel clinical phenotype.
Zhongguo Dang Dai Er Ke Za Zhi. 2009a;11:328–32. [
PubMed: 19470249]
Song YZ, Hao H, Ushikai M, Liu GS, Xiao X, Saheki T, Kobayashi K, Wang ZN. A difficult and complicated case study: neonatal intrahepatic cholestasis caused by citrin deficiency.
Zhongguo Dang Dai Er Ke Za Zhi. 2006;8:125–8. [
PubMed: 16613706]
Song YZ, Li BX, Chen FP, Liu SR, Sheng JS, Ushikai M, Zhang CH, Zhang T, Wang ZN, Kobayashi K, Saheki T, Zheng XY. Neonatal intrahepatic cholestasis caused by citrin deficiency: clinical and laboratory investigation of 13 subjects in mainland of China.
Dig Liver Dis. 2009b;41:683–9. [
PubMed: 19185551]
Song YZ, Sheng JS, Ushikai M, Hwu WL, Zhang CH, Kobayashi K. Identification and diagnosis of three novel mutations in SLC25A13 gene of neonatal intrahepatic cholestasis caused by citrin deficiency.
Zhonghua Er Ke Za Zhi. 2008;46:411–5. [
PubMed: 19099775]
Song YZ, Wen F, Chen FP, Kobayashi K, Saheki T. Neonatal intrahepatic cholestasis caused by citrin deficiency: efficacy of therapeutic formulas and update of clinical outcomes. Jpn J Inherit Metab Dis. 2010;26:57–69.
Song YZ, Zhang ZH, Lin WX, Zhao XJ, Deng M, Ma YL, Guo L, Chen FP, Long XL, He XL, Sunada Y, Soneda S, Nakatomi A, Dateki S, Ngu LH, Kobayashi K, Saheki T.
SLC25A13 gene analysis in citrin deficiency: sixteen novel mutations in Asian patients, and the mutation distribution in a large pediatric cohort in China.
PLoS One. 2013;8:e74544. [
PMC free article: PMC3777997] [
PubMed: 24069319]
Tabata A, Sheng J-S, Ushikai M, Song Y-Z, Gao H-Z, Lu Y-B, Okumura F, Iijima M, Mutoh K, Kishida S, Saheki T, Kobayashi K. Identification of 13 novel mutations including a retrotransposal insertion in SLC25A13 gene and frequency of 30 mutations found in patients with citrin deficiency.
J Hum Genet. 2008;53:534–45. [
PubMed: 18392553]
Takagi H, Hagiwara S, Hashizume H, Kanda D, Sato K, Sohara N, Kakizaki S, Takahashi H, Mori M, Kaneko H, Ohwada S, Ushikai M, Kobayashi K, Saheki T. Adult onset type II citrullinemia as a cause of non-alcoholic steatohepatitis.
J Hepatol. 2006;44:236–9. [
PubMed: 16278034]
Takahashi H, Kagawa T, Kobayashi K, Hirabayashi H, Yui M, Begum L, Mine T, Takagi S, Saheki T, Shinohara Y. A case of adult-onset type II citrullinemia - deterioration of clinical course after infusion of hyperosmotic and high sugar solutions.
Med Sci Monit. 2006;12:CS13–5. [
PubMed: 16449956]
Takahashi Y, Koyama S, Tanaka H, Arawaka S, Wada M, Kawanami T, Haga H, Watanabe H, Toyota K, Numakura C, Hayasaka K, Kato T. An elderly Japanese patient with adult-onset type II citrullinemia with a novel D493G mutation in the SLC25A13 gene.
Intern Med. 2012;51:2131–4. [
PubMed: 22892490]
Takaya J, Kobayashi K, Ohashi A, Ushikai M, Tabata A, Fujimoto S, Yamato F, Saheki T, Kobayashi Y. Variant clinical courses of 2 patients with neonatal intrahepatic cholestasis who have a novel mutation of SLC25A13.
Metabolism. 2005;54:1615–9. [
PubMed: 16311094]
Takeuchi S, Yazaki M, Yamada S, Fukuyama T, Inui A, Iwasaki Y, Ikeda S (2015) An adolescent case of citrin deficiency with severe anorexia mimicking anorexia nervosa. Pediatrics136:e530-4. [
PubMed: 26195537]
Tamakawa S, Nakamura H, Katano T, Yoshizawa M, Ohtake K, Kubota T. Hyperalimentation therapy produces a comatose state in a patient with citrullinemia. J Jpn Soc Intensive Care Med. 1994;1:37–41.
Tamamori A, Fujimoto A, Okano Y, Kobayashi K, Saheki T, Tagami Y, Takei H, Shigematsu Y, Hata I, Ozaki H, Tokuhara D, Nishimura Y, Yorifuji T, Igarashi N, Ohura T, Shimizu T, Inui K, Sakai N, Abukawa D, Miyakawa T, Matsumori M, Ban K, Kaneko H, Yamano T. Effects of citrin deficiency in the perinatal period: feasibility of newborn mass screening for citrin deficiency.
Pediatr Res. 2004;56:608–14. [
PubMed: 15295082]
Tamamori A, Okano Y, Ozaki H, Fujimoto A, Kajiwara M, Fukuda K, Kobayashi K, Saheki T, Tagami Y, Yamano T. Neonatal intrahepatic cholestasis caused by citrin deficiency: severe hepatic dysfunction in an infant requiring liver transplantation.
Eur J Pediatr. 2002;161:609–13. [
PubMed: 12424587]
Tanaka T, Nagao M, Tsutsumi H. Application of mutation analysis for the previously uncertain cases of adult-onset type II citrullinemia (CTLN2) and their clinical profiles.
Tohoku J Exp Med. 2002;198:89–97. [
PubMed: 12512993]
Tazawa Y, Abukawa D, Sakamoto O, Nagata I, Murakami J, Iizuka T, Okamoto M, Kimura A, Kurosawa T, Iinuma K, Kobayashi K, Saheki T, Ohura T. A possible mechanism of neonatal intrahepatic cholestasis caused by citrin deficiency.
Hepatol Res. 2005;31:168–71. [
PubMed: 15777702]
Tokuhara D, Iijima M, Tamamori A, Ohura T, Takaya J, Maisawa S, Kobayashi K, Saheki T, Yamano T, Okano Y. Novel diagnostic approach to citrin deficiency: Analysis of citrin protein in lymphocytes.
Mol Genet Metab. 2007;90:30–6. [
PubMed: 17092749]
Tomomasa T, Kobayashi K, Kaneko H, Shimura H, Fukusato T, Tabata M, Inoue Y, Ohwada S, Kasahara M, Morishita Y, Kimura M, Saheki T, Morikawa A. Possible clinical and histologic manifestations of adult-onset type II citrullinemia in early infancy.
J Pediatr. 2001;138:741–3. [
PubMed: 11343053]
Tong F, Yang JB, Huang XL, Zhou XL, Yang RL. A case of neonatal intrahepatic cholestasis caused by citrin deficiency complicated with congenital biliary atresia.
Zhonghua Er Ke Za Zhi. 2013;51:863–5. [
PubMed: 24484564]
Tsai C-W, Yang C-C, Chen H-L, Hwu W-L, Wu M-Z, Liu K-L, Wu M-S. Homozygous SLC25A13 mutation in a Taiwanese patient with adult-onset citrullinemia complicated with steatosis and hepatocellular carcinoma.
J Formos Med Assoc. 2006;105:852–6. [
PubMed: 17000460]
Tsuboi Y, Fujino Y, Kobayashi K, Saheki T, Yamada T. High serum pancreatic secretory trypsin inhibitor before onset of type II citrullinemia.
Neurology. 2001;57:933. [
PubMed: 11552040]
Wang H, Shu S, Chen C, Huang Z, Wang D. Novel mutations in the SLC25A13 gene in a patient with NICCD and severe manifestations.
J Pediatr Endocrinol Metab. 2015;28:471–5. [
PubMed: 25381944]
Wen PQ, Wang GB, Chen ZL, Liu XH, Cui D, Shang Y, Li CR. Clinical investigation and mutation analysis of a child with citrin deficiency complicated with purpura, convulsive seizures and methioninemia.
Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2013;30:649–53. [
PubMed: 24327139]
Wong LJ, Dimmock D, Geraghty MT, Quan R, Lichter-Konecki U, Wang J, Brundage EK, Scaglia F, Chinault AC. Utility of oligonucleotide array-based comparative genomic hybridization for detection of target gene deletions.
Clin Chem. 2008;54:1141–8. [
PubMed: 18487280]
Xing YZ, Qiu WJ, Ye J, Han LS, Xu SS, Zhang HW, Gao XL, Wang Y, Gu XF. Studies on the clinical manifestation and SLC25A13 gene mutation of Chinese patients with neonatal intrahepatic cholestasis caused by citrin deficiency.
Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2010;27:180–5. [
PubMed: 20376801]
Xiong XL, Yan SQ, Ding Y, Zhou LS, Chen P. Clinical research of neonatal intrahepatic cholestasis caused by citrin deficiency in Hubei Province. Ch J Appl Clin Pediatr. 2015;14:1064–8. ZHao DC.
Yamaguchi N, Kobayashi K, Yasuda T, Nishi I, Iijima M, Nakagawa M, Osame M, Kondo I, Saheki T. Screening of SLC25A13 mutations in early and late onset patients with citrin deficiency and in the Japanese population: Identification of two novel mutations and establishment of multiple DNA diagnosis methods for nine mutations.
Hum Mutat. 2002;19:122–30. [
PubMed: 11793471]
Yasuda T, Yamaguchi N, Kobayashi K, Nishi I, Horinouchi H, Jalil MA, Li MX, Ushikai M, Iijima M, Kondo I, Saheki T. Identification of two novel mutations in the SLC25A13 gene and detection of seven mutations in 102 patients with adult-onset type II citrullinemia.
Hum Genet. 2000;107:537–45. [
PubMed: 11153906]
Yazaki M, Hashikura Y, Takei Y, Ikegami T, Miyagawa S, Yamamoto K, Tokuda T, Kobayashi K, Saheki T, Ikeda S. Feasibility of auxiliary partial orthotopic liver transplantation from living donors for patients with adult-onset type II citrullinemia.
Liver Transpl. 2004;10:550–4. [
PubMed: 15048800]
Yazaki M, Ikeda S, Kobayashi K, Saheki T. Therapeutic approaches for patients with adult-onset type II citrullinemia (CTLN2): effectiveness of treatment with low-carbohydrate diet and sodium pyruvate.
Rinsho Shinkeigaku. 2010;50:844–7. [
PubMed: 21921468]
Yazaki M, Takei Y, Kobayashi K, Saheki T, Ikeda S. Risk of worsened encephalopathy after intravenous glycerol therapy in patients with adult-onset type II citrullinemia (CTLN2).
Intern Med. 2005;44:188–95. [
PubMed: 15805705]
Yeh JN, Jeng YM, Chen HL, Ni YH, Hwu WL, Chang MH. Hepatic steatosis and neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) in Taiwanese infants.
J Pediatr. 2006;148:642–6. [
PubMed: 16737877]
Zeng HS, Lin WX, Zhao ST, Zhang ZH, Yang HW, Chen FP, Song YZ, Yin ZN.
SLC25A13c DNA cloning analysis using peripheral blood lymphocytes facilitates the identification of a large deletion mutation: Molecular diagnosis of an infant with neonatal intrahepatic cholestasis caused by citrin deficiency.
Mol Med Rep. 2016;14:5189–94. [
PubMed: 27779681]
Zeng HS, Zhao ST, Deng M, Zhang ZH, Cai XR, Chen FP, Song YZ. Inspissated bile syndrome in an infant with citrin deficiency and congenital anomalies of the biliary tract and esophagus: Identification and pathogenicity analysis of a novel
SLC25A13 mutation with incomplete penetrance.
Int J Mol Med. 2014;34:1241–8. [
PMC free article: PMC4199400] [
PubMed: 25216257]
Zhang ZH, Lin WX, Deng M, Zhao ST, Zeng HS, Chen FP, Song YZ. Clinical, molecular and functional investigation on an infant with neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD).
PLoS One. 2014a;9:e89267. [
PMC free article: PMC3931723] [
PubMed: 24586645]
Zhang ZH, Lin WX, Zheng QQ, Guo L, Zhou QH, Song YZ. Molecular diagnosis of citrin deficiency in an infant with intrahepatic cholestasis: identification of a 21.7kb gross deletion that completely silences the transcriptional and translational expression of the affected SLC25A13 allele.
Oncotarget. 2017 [
PMC free article: PMC5675625] [
PubMed: 29152073]
Zhang ZH, Yang ZG, Chen FP, Kikuchi A, Liu ZH, Kuang LZ, Li WM, Song YZ, Kure S, Saheki S. Screening for five prevalent mutations of
SLC25A13 gene in Guangdong, China: A molecular epidemiological survey of citrin deficiency.
Tohoku J Exp Med. 2014b;233:275–81. [
PubMed: 25110155]
Zheng QQ, Zhang ZH, Zeng HS, Lin WX, Yang HW, Yin ZN, Song YZ. Identification of a large
SLC25A13 deletion via sophisticated molecular analyses using peripheral blood lymphocytes in an infant with Neonatal Intrahepatic Cholestasis caused by Citrin Deficiency (NICCD): A clinical and molecular study.
Biomed Res Int. 2016;2016:4124263. [
PMC free article: PMC4835617] [
PubMed: 27127784]
Chapter Notes
Author Notes
The first author of this review, Keiko Kobayashi, PhD, died of colon cancer on December 21, 2010. The scientific community has lost a great scientist, teacher, and friend.
Keiko Kobayashi is recognized internationally as a pioneer in citrin deficiency research. As an investigator in the research group of Professor Takeyori Saheki (Department of Molecular Metabolism and Biochemical Genetics, Kagoshima University, Japan), in 1999 she cloned the causative gene SLC25A13 and designated the term "citrin" for citrin deficiency. Kobayashi also played essential roles in the discovery and designation of NICCD and FTTDCD, two early-onset forms of citrin deficiency. As an outstanding molecular geneticist, she identified more than 50 pathogenic variants in SLC25A13 and diagnosed more than 500 citrin-deficient patients worldwide(Japan, Korea, China, Vietnam, Malaysia, Israel, Palestine, Australia, Czech, France, Britain, and the US). She also worked tirelessly to educate the medical community about citrin deficiency, thus improving the care and prognosis of affected patients worldwide. Less than a month before her death, Dr Kobayashi delivered a lecture on citrin deficiency to the 9th Asia-Pacific Conference on Human Genetics.
Keiko Kobayashi, the "mother of citrin deficiency," will be remembered and sorely missed by her friends, students, colleagues, and the citrin-deficient patients whom she diagnosed.
Acknowledgments
This research was supported in part by Grants-in-Aid for Scientific Research (Nos. 16390100, 19390096 and 19591230) and for Asia-Africa Scientific Platform Program (AASPP) from the Japan Society for the Promotion of Science (JSPS), by a Grant for Child Health and Development (17-2) from the Ministry of Health, Labour and Welfare in Japan, by a Grant for Research for Promoting Technological Seeds from the Japan Science and Technology Agency and by the Projects 81070279,81270957 and 81570793 supported by the National Natural Science Foundation (NSFC) of China.
Author History
Keiko Kobayashi, PhD; Kagoshima University Graduate School of Medical and Dental Sciences, Kagoshima, Japan (2004-2010)
Takeyori Saheki, MD, PhD (2004-present)
Yuan-Zong Song, MD, PhD (2012-present)
Revision History
10 August 2017 (ha) Comprehensive update posted live
31 July 2014 (me) Comprehensive update posted live
5 January 2012 (me) Comprehensive update posted live
1 July 2008 (me) Comprehensive update posted live
16 September 2005 (me) Review posted live
5 November 2004 (kk) Original submission