Clinical Description
Aicardi syndrome, first described by Aicardi et al [1965], is a neurodevelopmental disorder that affects primarily females [Aicardi 1999, Van den Veyver 2002, Aicardi 2005]. Initially it was characterized by a typical triad of agenesis of the corpus callosum, typical chorioretinal lacunae, and infantile spasms; however, as more affected individuals have been ascertained, it has become clear that other neurologic and systemic defects are common. Indeed, not all affected girls have all three features of the classic triad (see Table 1).
Table 1.
Select Features of Aicardi Syndrome
View in own window
| Feature | % of Persons
w/Feature | Comment |
|---|
|
Neurologic
| Seizures | >95% | Nearly all present w/infantile spasms in 1st mos of life; various other seizure types develop over time. |
| ID/DD | 100% | Many have severe-profound ID & do not develop language or ability to walk or sit independently; however, the degree of developmental impairment varies. |
Structural brain
malformations | 100% | While agenesis of the corpus callosum is part of the classic triad, virtually all have other brain malformations (e.g., heterotopias, cysts). |
|
Ocular
| Chorioretinal
lacunae | 100% | Typically in central part of retina (not periphery); may cause visual impairment if they involve the macula; may be bilateral or unilateral |
Optic nerve
abnormalities | >90% | Colobomas & hypoplasia of optic nerves are common & → visual impairment. |
|
Other
| Costovertebral
abnormalities | ~50% | Fusion of vertebrae & ribs can be seen, often (along w/severe hypotonia) → scoliosis. |
DD = developmental delay; ID = intellectual disability
Neurologic. The neurologic examination can reveal microcephaly, axial hypotonia, and appendicular hypertonia with spasticity often affecting one side and brisk deep tendon reflexes as well as hemiparesis [Aicardi 2005; Author, unpublished data]. Moderate-to-severe developmental delay and intellectual disability are typical, although individuals with only mild or no learning disabilities or developmental delay have been reported (reviewed in Wong & Sutton [2018]).
Many girls with Aicardi syndrome develop seizures before age three months, and most before age one year. Infantile spasms are seen early on; ongoing medically refractory epilepsy with a variety of seizure types develops over time. Common EEG findings include asynchronous multifocal epileptiform abnormalities with burst suppression and dissociation between the two hemispheres.
Brain MRI reveals dysgenesis of the corpus callosum, which is most often complete but can be partial. Polymicrogyria or pachygyria, which are predominantly frontal and perisylvian and associated with underopercularization, are typical. Periventricular and intracortical gray matter heterotopia are very common. Gross cerebral asymmetry, choroid plexus papillomas, ventriculomegaly, and intracerebral cysts, often at the third ventricle and in the choroid plexus, are frequently present [Aicardi 2005, Hopkins et al 2008]. Recently, posterior fossa and cerebellar abnormalities are increasingly recognized as important components of the phenotype [Hopkins et al 2008, Steffensen et al 2009].
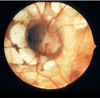
Ophthalmologic. The pathognomonic chorioretinal lacunae of Aicardi syndrome are white or yellow-white, well-circumscribed, round, depigmented areas of the retinal pigment epithelium and underlying choroid with variably dense pigmentation at their borders () [Fruhman et al 2012] that can cluster in the posterior pole of the globe around the optic nerve. The sensory retina overlying the lacunae is usually intact but may be disorganized or entirely absent.
Classic lacunae surround the modestly dysplastic left optic disc. Note the nasal "papilla nigra" appearance and the anomalous branching patterns of the central vasculature. Image courtesy of Dr Richard A Lewis
Almost all individuals have some ocular abnormalities. Fruhman et al [2012] described data on 40 females with Aicardi syndrome examined by a single expert ophthalmologist and retinal specialist. Of the 75 eyes that could be evaluated, all had chorioretinal lacunae. Other common optic nerve findings included:
Coloboma (39% of eyes)
Glial proliferation (19% of eyes)
Severe dysplasia (17% of eyes)
Pseudoadenomatous proliferation of the retinal pigment epithelium (14% of eyes)
All eye findings can be unilateral or bilateral and asymmetric.
Craniofacial. Characteristic facial features reported in Aicardi syndrome include a short philtrum, prominent premaxilla with resultant upturned nasal tip and decreased angle of the nasal bridge, large ears, and sparse lateral eyebrows [Sutton et al 2005].
Plagiocephaly and facial asymmetry, occasionally with cleft lip and palate (3%), have been reported. Pierre Robin sequence has been reported in a single case [Jensen & Christiansen 2004].
Skeletal. Costovertebral defects, such as hemivertebrae, block vertebrae, fused vertebrae, and missing ribs, are common and can lead to marked scoliosis in up to one third of affected individuals [Donnenfeld et al 1989]. Hip dysplasia has been reported.
Gastrointestinal. Constipation, gastroesophageal reflux, diarrhea, and feeding difficulties are perceived by parents to be the second most difficult problem to manage (after seizures) [Glasmacher et al 2007].
Extremities. Small hands, along with an increased incidence of hand malformations, have been reported [Sutton et al 2005].
Dermatologic. An increased incidence of vascular malformations and pigmentary lesions has been observed [Sutton et al 2005].
Tumors/malignancies. The incidence of tumors may be increased. A variety of rare tumor types have been reported: benign tumors such as choroid plexus papillomas [Taggard & Menezes 2000, Pianetti Filho et al 2002] and lipomas, as well as malignant tumors including angiosarcomas, hepatoblastomas, meduloblastoma, embryonal carcinomas, and teratomas [Sutton et al 2005, Wharton et al 2018].
Growth. The average heights and weights of girls with Aicardi syndrome closely follow those of the general population up to ages seven and nine years, respectively, after which the growth rate for both height and weight is lower. Growth curves for Aicardi syndrome based on parent survey data have been published [Glasmacher et al 2007]. The weight-versus-height ratio remains similar to the general population.
Endocrine. Either precocious puberty or delayed puberty may be present [Glasmacher et al 2007].
Survival. Survival in Aicardi syndrome is highly variable and likely depends on the severity of seizures. In a survey by Glasmacher et al [2007], the mean age at death was 8.3 years, although the median age of death was 18.5 years. The ages of death were distributed from before age one year to older than 23 years. The oldest surviving individual reported in this survey was age 32 years. Another paper also indicated that the survival is longer than previously reported, especially in more mildly affected individuals, with the highest risk of death at age 16 years and a probability of survival at age 27 years of 0.62 [Kroner et al 2008].