This publication is provided for historical reference only and the information may be out of date.
Scope
The NCBI Clone DB is a database that integrates information about eukaryotic genomic and cell-based clones and libraries, including sequence data, genomic location, and distribution sources (1). At Clone DB, users can find library metadata, search for clones containing genes or other sequences of interest, or find contact information for distributors of libraries and clones. In addition, Clone DB provides mapping data that can be used to help researchers assess and improve genome assemblies. Although Clone DB is a resource whose aim is to help users connect data with physical clone reagents, NCBI is not itself a distributor of libraries or clones. The database contains library and clone records for over 150 taxa, is indexed in Entrez, and can be searched by many terms, including clone, library or gene name, organism or sequence identifier. Clone DB maps genomic clones to reference assemblies when such data is available. These placements can be viewed as graphical displays in the clone records themselves, as well as in the NCBI Clone Finder, where clone placements can be searched by location, genome features, or transcript names.
Clone DB maintains records for genomic and cell-based libraries and clones that are available from commercial or academic distributors, along with a limited collection of clone libraries of sufficient scientific significance to warrant their representation even in the absence of distribution. At this time, Clone DB contains over five hundred genomic library records that represent more than 150 different eukaryotic taxa, which include both animal and plant species. The current Clone DB collection of records for cell-based clones includes gene trap and gene target libraries produced by the International Knock-out Mouse Consortium (IKMC) (2, 3) and International Gene Trap Consortium (IGTC) (4), as well as the Lexicon Genetics gene trap collection (5). These libraries and their associated metadata are provided to Clone DB by Mouse Genome Informatics (MGI). Genomic library records in Clone DB include the original set of libraries imported from the former NCBI Clone Registry database, as well as additional library records generated by database curators. Curators continue to update the database with new library records, emphasizing representation for genomic libraries that contribute to the generation of reference assemblies, are extensively end or insert sequenced or fingerprinted, as well as libraries whose representation is specifically requested by users contacting the Clone DB (vog.hin.mln.ibcn@nimda-gerenolc).
History
Clone DB replaces and extends upon the former NCBI Clone Registry. The Clone Registry was developed during the Human Genome Project (HGP) as a resource to assist the many large-scale sequencing centers involved in this effort track the sequencing of clones in the tiling paths for the human and mouse reference assemblies. Importantly, the Clone Registry developed the notion of a standardized clone naming system that could be used to unambiguously identify clones from different libraries. This naming system was adopted by many of the sequencing centers and facilitated the consolidation of clone data from various and disparate databases. Clone Registry records contained information about sequencing status, links to end and insert sequence records, mapping locations, and clone distributors. Although the Clone Registry later grew to include records for genomic clones from other eukaryotic taxa, these generally lacked the depth of the human and mouse records, because of the relative paucity of genomic data. As the HGP drew to a close, it was clear that the Clone Registry would need to evolve in order to remain a relevant resource.
Clones continue to play an important role in biological research in the current era of next generation sequencing technologies, though their specific uses have changed. Although whole genome sequencing (WGS) has largely obviated the use of clone tiling paths in the generation of genome assemblies, genomic clones are still among the best means for resolving sequences in complex regions. Clone end alignments are used to assess assembly quality, and the technique of end-sequence profiling has proven to be a valuable means for discovering genomic variation (6, 7). In organisms in which large amounts of repetitive content or variation confound WGS assemblies, genomic clones remain the sequencing reagent of choice (8). Cell-based clones, such as gene trap and gene targeting clones, are used in the study of many model organisms to define gene function and study genotype-phenotype relationships (2, 9-11). In all these instances, clones may be associated with a variety of data types. Thus, there remains a need for a clone-focused database that can consolidate this information and assist users in obtaining these important biological reagents. Clone DB now serves this function, providing users with a central location to access information about library construction details and clone sequence, gene content, map location, and distribution.
Data Model
The data objects in Clone DB represent physical objects. Thus, the physical attributes of clones and libraries inform the Clone DB data model (Figure 1).
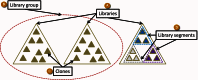
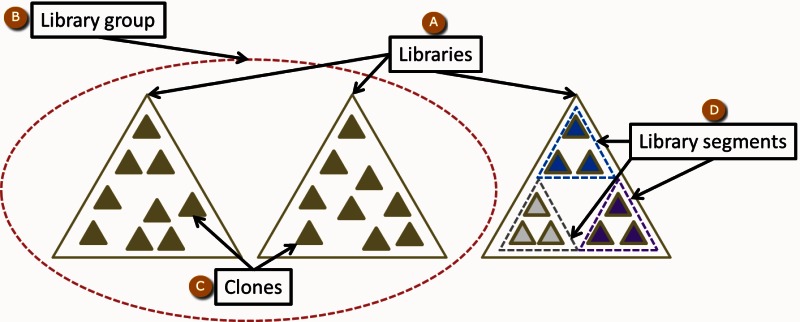
Libraries
Functionally speaking, a library is a collection of clones, and Clone DB utilizes this hierarchical relationship in its data model. In Clone DB, all clone records must be associated with a library record. Within a species, each library is uniquely identified in Clone DB by its library name. Metadata collected for genomic clone libraries includes details about source DNA, library construction, library statistics, alternate names and abbreviations by which the library is known, and library distributors, as well as publications describing the construction or end sequencing of the library. Metadata captured for cell-based libraries includes the library creator, cloning vector, parental cell line, parental cell line strain, and allele type.
Library Groups
Within the database, library groups may be defined for collections of libraries that share one or more common features. Any common feature may be used as the basis for the creation of a library group. For example, murine cell-based libraries generated as part of the International Knock-Out Mouse Consortium (IKMC) belong to the same library group.
Library Segments
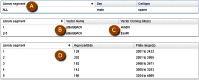
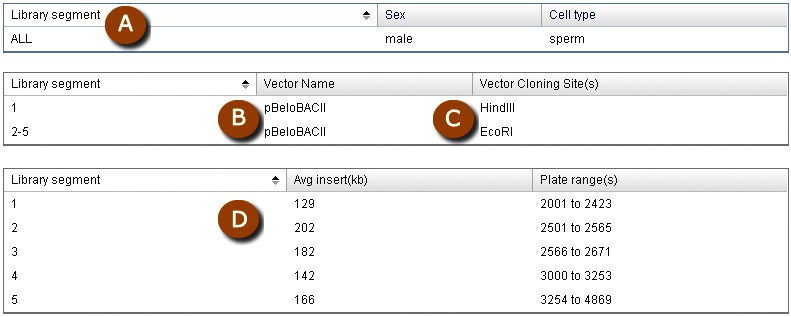
Within a single library, there may be subsets of clones that are distinguished by different sets of common attributes. Such attributes may include, but are not restricted to, cloning vector, vector type, source DNA, or average insert size. Clone DB uses the notion of library segments to capture any such subsets within a library record. Displays for library records report both the features common to the library as a whole, as well as segmental differences. For example, the record for the Caltech Human BAC Library D in Clone DB has 5 segments (Figure 2). All of the segments share a common DNA source and cloning vector, but the DNA for clones in one segment were digested by and cloned into the site for a different restriction enzyme. The 5 segments of this library, which were defined by the library creators, are distinguished by different average insert sizes. Within the physical library, these segments correspond to different sets of numbered microtiter dishes (plate ranges).
Clones
Clone DB also includes records for individual clones. As noted above, all clone records must be associated with a library record. Although library records are representations of physical libraries, Clone DB does not necessarily maintain records for all of the physical clones that are associated with a particular physical library. Instead, records are only created for those clones for which there are sequence or mapping data to be represented in Clone DB.
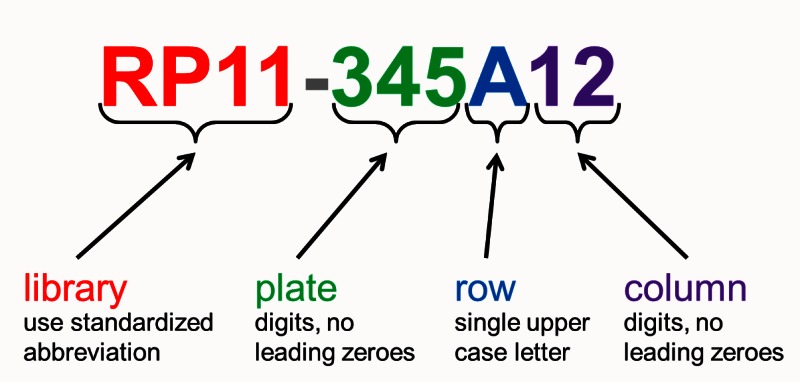
A major feature of each clone’s record is the clone name assigned by Clone DB. The assignment of names to genomic clones often presents a challenge, as it is common to find that different submitters have provided different permutations of a clone name on different data submissions representing the same clone object. Whenever possible, Clone DB attempts to parse submitter-provided names and assign a standardized name comprised of the clone’s microtiter plate address (plate number, row and column), prefixed by a Clone DB library abbreviation to each record (Figure 3). In such cases, the submitter-provided name will be stored as a searchable alias of the standard name. If a standard name cannot be parsed from the submitter-provided name, the submitter-provided name will be assigned as the clone name. All names and aliases associated with genomic clone records are indexed and can be used as search queries. In the case of murine cell-based clones, Clone DB simply adopts the clone name provided by MGI.
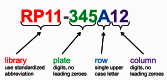
Figure 3.
Clone DB standard nomenclature for genomic clones. Standardized library abbreviations are unique for each species in Clone DB.
Dataflow
Record Data
Metadata associated with records in Clone DB are supplied by different sources. On a weekly basis, MGI provides the clone library, gene, allele, and sequence identifier information, as well as creator and distributor details, for all murine cell-based clone records in Clone DB. In contrast, data affiliated with genomic clone records are derived from a variety of NCBI databases and external providers. Clone DB queries the Nucleotide database daily to retrieve high throughput genomic (HTG) insert sequences and their associated metadata for all organisms that have at least one library represented in the clone database. End sequences and their metadata are retrieved from both the GSS and Trace Archive databases via library-specific queries on an ad-hoc basis. Fingerprint data for genomic clones has been obtained from the FPC database maintained by the Michael Smith Genome Sciences Centre in Vancouver, Canada (http://www.bcgsc.ca/data/data) and The Genome Institute at Washington University, St. Louis. Likewise, data for cytogenetic map positions and STS markers mapped to human genomic clones are taken from the work of the BAC resource consortium (12) and National Cancer Institute’s Cancer Chromosome Aberration Project (13).
Record Curation
All new genomic library records are generated and loaded to Clone DB by database curators. Weekly queries produce reports that identify insert and end sequences without corresponding libraries for organisms already represented in Clone DB. Curators also perform literature reviews to identify additional organisms for which record creation may be needed. Furthermore, new library records may be added at the behest of library creators, distributors, and database users by contacting Clone DB (vog.hin.mln.ibcn@nimda-gerenolc). All queries to retrieve end sequences from the Trace Archives and GSS are also defined by curators. In addition, curation is performed to address issues with library and clone record metadata that are identified by automated processes that check for data integrity. For example, retrieved insert and end sequence records that contain clone names that cannot be associated with existing clone records, or from which new standardized clone names cannot be parsed and created, are flagged for curatorial review. Likewise, externally provided data receives curatorial review to ensure consistency with the Clone DB data model. End sequence and clone placements are also reviewed by curators with respect to size, number, and concordance to ensure that the NCBI clone placement algorithm is producing results consistent with published library and genome characteristics.
Genomic Clone Placements
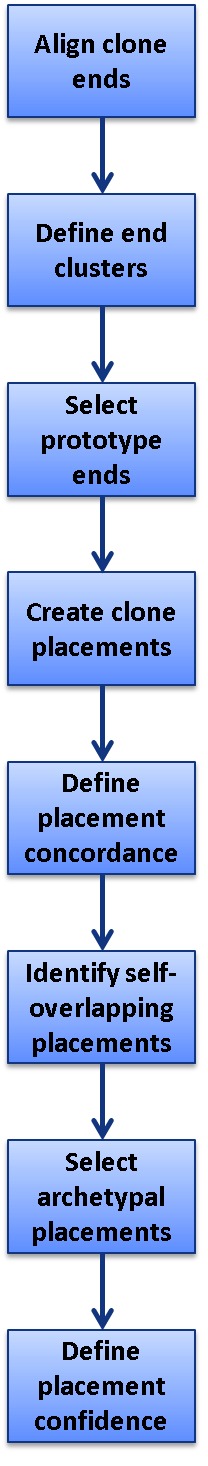
Clone DB maps genomic clones to assemblies that are annotated by the NCBI eukaryotic genome annotation pipeline. As a genomic clone is a physical object containing a specific fragment of DNA, this mapping provides context for the clone with respect to its genomic origin and to other clones in the same and other libraries. At the time of this handbook’s writing, the NCBI clone placement algorithm only uses end sequences to create clone placements. In the future, these placements may also be informed by insert sequences. End sequences associated with clone records are screened to remove vector contamination and low quality bases. The set of processed ends is aligned to the genome assembly of interest with NG Aligner, an NCBI BLAST-derived aligner (see Genome Workbench chapter). In most cases, the ends are aligned to a genome representing the same species as the clone library. However, in instances in which no such genome is available, ends may be mapped to the genome of a closely related species.
The data flow for the generation of clone placements by Clone DB is illustrated in Figure 4. Clone placements are created by pairing end placements representing opposite ends of the same clone. The NCBI clone placement algorithm uses two mechanisms to minimize self-overlapping clone placements and present users with the most likely placement(s) for each clone. First, the algorithm clusters any overlapping end placements for a given clone end and selects only a single prototype for use in clone placements. The prototype is the end placement that holds the 5’-outermost position on the scaffold to which it aligns. Second, if the end placement prototypes contribute to a set of self-overlapping clone placements, the algorithm uses a set of defined heuristics to select a single clone placement from the set as an archetypal placement. A clone may therefore have more than one archetypal placement, but these may not overlap. Only archetypal placements are reported and displayed in clone records.
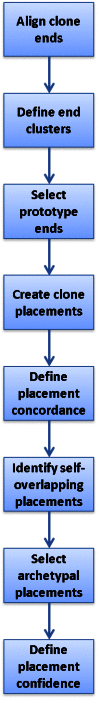
Figure 4.
Diagram illustrating data flow for genomic clone placements in Clone DB.
Clone DB defines an average insert size and standard deviation for each library based on its clone placements. It should be noted that these values, which are provided in reports on the Clone DB FTP site, may differ slightly from the library creator-provided values reported in the library record displays. As the latter values are commonly defined using techniques other than end mapping (e.g. gel sizing), some discrepancies are to be expected.
Clone DB defines the average insert size and standard deviation for each library using only the following subset of clone placements on an assembly:
- the placement is comprised of 1 forward and 1 reverse end
- both ends are uniquely placed
- both ends are placed on the same assembly scaffold
- end placements face each other
- placement length is between 50-500 kb (BAC/PAC) or 10-100 kb (fosmids).
Clone DB defines concordant placements as those in which:
- placement length is within 3 standard deviations of the library average
- contributing end placements are facing one another on opposite strands
- Any clone placement not meeting either of these criteria is defined as discordant. No placement-related data is provided for clones for which no placements are found.
Clone placements are also assigned a category reflecting the level of confidence in the placement. Confidence assignments are reported in the “Clone Placement” tab of individual genomic clone records.
- Unique: There is a unique placement for the clone within an assembly unit. All end placements associated with the clone support this placement.
- Unique-dissent: There is a unique placement for the clone within an assembly unit, but there are end placements that do not support the clone placement.
- Multiple-conflict: There are multiple placements for the clone in an assembly unit; every clone placement is comprised of two uniquely placed ends and the multiple placements are due to non-overlapping end clusters.
- Multiple-best: There are multiple placements for the clone in an assembly unit; this clone placement is comprised of two top-ranked end placements and the multiple placements are due to the existence of end sequences with multiple end placements.
- Multiple-other: There are multiple placements for the clone in an assembly unit; this clone placement is comprised of lower-ranked end placements and the multiple placements are due to the existence of end sequences with multiple end placements.
Additional details describing the clone placement process are provided in documentation on the Clone DB website.
Access
Data from Clone DB can be accessed via FTP, direct Entrez query, or through use of tools such as the Clone DB library browsers or Clone Finder.
FTP site
At the Clone DB FTP site, users will find a number of reports that provide details about clone-associated sequences and clone placements. These reports, which are organized by species, include:
- clone_acstate: details of genomic clone insert sequences (updated weekly)
- clone_placement_report: summary information for clone placements generated by Clone DB (updated whenever new placements are generated)
- endinfo: details of genome clone end sequences (updated weekly)
- end_placement_report: summary information for end sequence placements generated by Clone DB (updated whenever new placements are generated)
- library: summary details for all clone libraries (updated weekly)
The FTP site also contains text files with the clone placements themselves. There may also be additional text files available for some organisms, depending on data availability.
Entrez search
As an Entrez-indexed database, Clone DB can be directly queried by entering text into the search box found at the top of all Clone DB web pages. For assistance with the construction of complex text queries, users may click on the “Advanced” link located beneath the search box. Documentation describing the complete set of indexed terms is accessed by clicking on the “Help” link found on each Clone DB web page. Query results are presented in tabular format where each row provides a result summary and a link to the corresponding library or clone record page (Figure 5).
![Figure 5. . Screenshot of results returned from an Entrez query of Clone DB (("gene targeting"[Library Type]) AND mouse[Organism]).](/books/NBK153524/bin/Clone-Image005.gif)
Library browsers
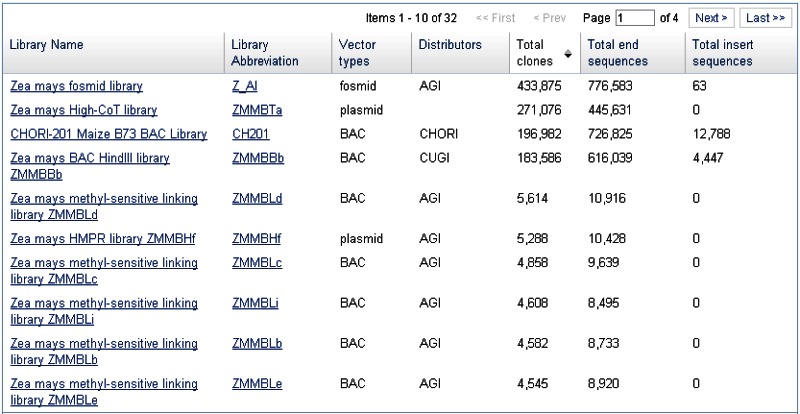
Clone DB also provides a pair of library browsers to facilitate user access to cell-based and genomic library records. These browsers, which are accessed via links on the Clone DB homepage, are sortable tables that provide summary information for each of the libraries represented in Clone DB (Figure 6). A set of filters can be used to restrict the displays to subsets of libraries meeting certain characteristics, such as organism, library or vector type, sequence count or distributor. Each row in the browser table provides a link to the corresponding library record. FAQ pages for the genomic and cell-based library browsers are provided to assist users with their navigation.
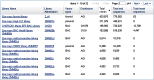
Clone DB Records
Libraries
Users can view cell-based and genomic library details on Clone DB’s individual library record pages. Library records can be accessed via links in the “Library Name” and “Library Abbreviation” columns in the genomic and cell-based library browsers, as well as from the “Library Name” column of the table in which results from an Entrez search of Clone DB are displayed. At the top of each library record page, a summary provides easy access to key library attributes, including library name, library group, organism and counts of the number of clones and associated sequences in the database. For genomic library records, this summary also includes distributor information. For cell-based library records, the summary provides the allele type. Below the summary, a tabbed table provides details about the library’s DNA source, construction, statistics, and aliases by which it is known (Figure 2). In the case of cell-based libraries, the table also provides information about the library host. On these pages, users will also find a link to the results of an Entrez query that returns the records for all clones in the library, as well as links that direct them to publications describing library construction and/or sequencing. The data presented in these records are intended to help researchers determine whether clones from the library will be suitable for their research needs and to facilitate their use in a research setting. For more information about these pages, please see the Clone DB Help page.
Clones
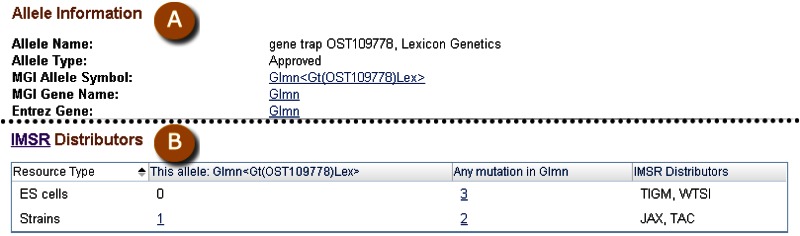
Details for individual clones can be viewed in Clone DB’s individual clone record pages, which can be accessed by performing an Entrez query of Clone DB and clicking on links in the “Clone Name” column of the table in which search results are displayed. Similar to the library record pages, the clone record pages also contain a summary section that presents a digest of key attributes. These include the standardized clone name, along with any aliases by which the clone is known, the library to which it belongs, and the library type. A tabbed table beneath the summary provides additional information about the clone. A tab specific to the murine cell-based clone record pages presents allele information, including type, name, and links to corresponding gene records at MGI and the NCBI Gene database (Figure 7). Cell-based and genomic clone records both have tabs that provide distribution details and information about corresponding sequences. In genomic clone records, users will find additional tabs containing data about genetic markers mapped to the clone, fingerprint contigs to which the clone belongs, and clone placement details (see below for more information about accessing clone placements). Detailed descriptions of the clone record pages can be found on the Clone DB Help page.

Clone Placements
Clone Records
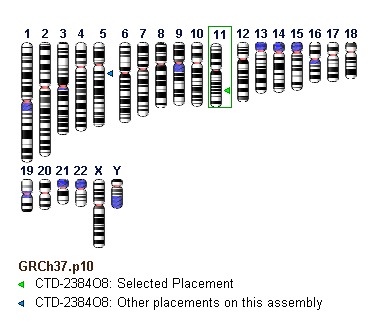
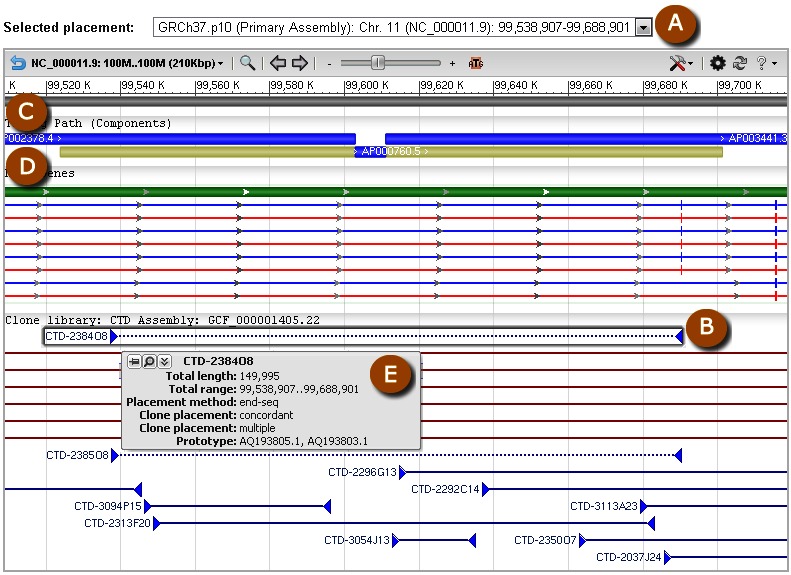
Graphical displays of NCBI clone placements can be accessed in individual clone records. Within Entrez, users can search for clones having placements using the query "placed"[Properties], and can also query to identify clones whose placements belong to any of the above-noted confidence categories by using the search field [Placement Confidence]. An ideogram at the top of each record page provides genomic context for any placements the clone may have (Figure 8). The “Genome View” tab displays an instance of the NCBI Sequence Viewer showing the placement of the clone in the context of other clones in the same library (Figure 9). If a clone has multiple placements, users can select the specific placement to be displayed. The selected placement for the clone of record is highlighted and shown at top. This display also shows assembly components and NCBI annotated genes. A FAQ page for clone placements provides a legend that explains the rendering scheme used in this graphical display. Holding the mouse over any placement will bring up a tool tip that includes additional placement details, including the concordance and uniqueness, as well as the sequence identifiers of the prototype ends that contributed to the placement. Additional placement information for the clone, including any non-sequence based placements (i.e., cytological), is provided in tabular format in the “Clone Placements” tab (Figure 10).

Figure 8:
Ideogram from individual genomic clone record displays showing clone placements (arrowheads).


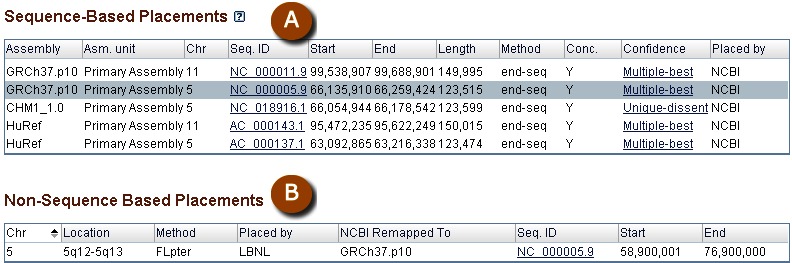
Figure 10.
Tabular placement displays from individual genomic library record page. A: Details for sequence-based clone placements. B: Details for non-sequence based clone placements.
Clone Finder
Clone placements may also be viewed with the NCBI Clone Finder resource, which can be accessed from the Clone DB home page. While the individual clone record pages enable users to review the placement details for a specific clone in the context of other clones from the same library, Clone Finder allows users to search for and visualize placements of clones from different libraries that have been mapped to specific genomic regions. Users may perform Clone Finder searches based on genomic location or feature, such gene or transcript name, marker or SNP. Filters are available to restrict searches by DNA source, library, or vector type (Figure 11). In contrast to the placement displays provided in individual clone records, Clone Finder can simultaneously display the placements for clones from different libraries. The Clone Finder graphical display distinguishes concordant and discordant placements and includes assembly components and annotated genes in the selected region (Figure 12). The placement data is also displayed in a tabular format (Figure 13) and can be downloaded in Excel.
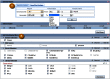
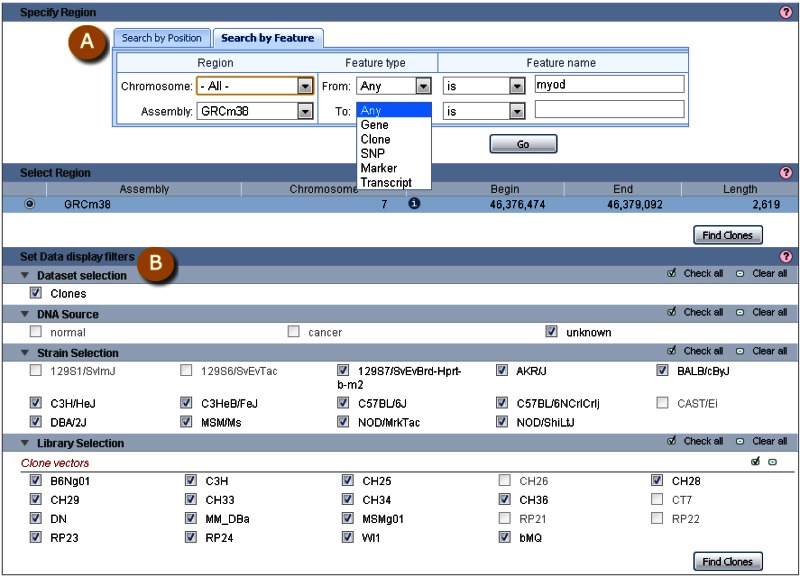
Figure 11.
Clone Finder search interface. A: Users may search for clone placements by chromosome coordinate or genomic feature. B: A number of filters allow users to restrict the display of placed clones.

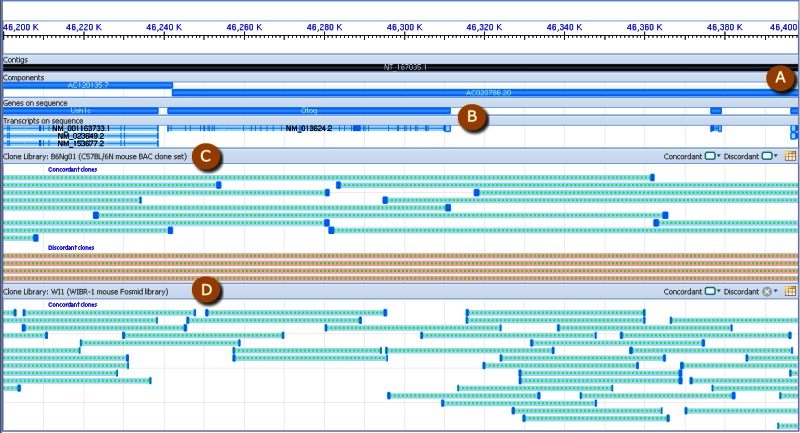
Figure 12.
Screenshot of Clone Finder graphical display. A: Assembly scaffolds and components. B: Gene and transcript annotation. C, D: Clone placements from two different libraries are shown. Concordant placements are in green, discordant placements are red.

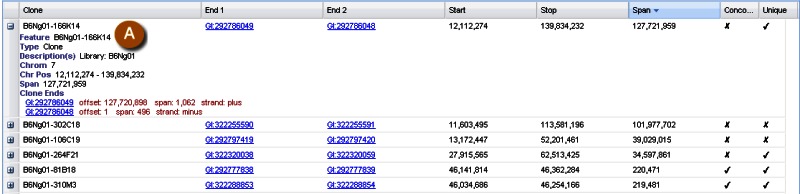
Figure 13.
Screenshot of Clone Finder tabular display. A: One of the rows has been expanded to show additional placement details.
Related Tools
Several related tools at NCBI are available that may be of interest to users of Clone DB.
- The Map Viewer “Clone” track presents clone placements generated by Clone DB.
- Only clones with concordant placements are displayed in this track.
- Utilities for accessing end sequence records
- endseq_dp.pl
- This is a perl script provided by Clone DB that dumps FASTA files for end sequences with records in dbGSS or Trace Archives
- It takes a list of NCBI GI numbers (max 1000) or Trace Archive identifiers (max 4000) as input and returns the corresponding FASTA sequences.
- The script and usage directions are located in the utility directory of the Clone DB FTP site.
- query_tracedb.pl
- This is a perl script provided by the Trace Archive that can be used to download large datasets from the Trace Archive.
- The script and usage directions are located in the “Obtaining Data” tab on the Trace Archive home page.
- End sequence BLAST databases
- NCBI BLAST databases comprised of end sequences from individual or collections of genomic clone libraries are available for several organisms, including human and mouse.
- These databases are listed in in the “Database” drop-down menu on organism-specific BLAST pages, the complete list of which can be accessed via the Map Viewer home page.
- Unless otherwise noted, BLAST databases named “Clone end sequences” only contain end sequences whose records are in dbGSS, not the Trace Archive.
- Genome Reference Consortium (GRC) annotated clone assembly problems files
- Available for human, mouse and zebrafish clones that are components of the respective reference assemblies for each of these organisms; these files map individual clone assembly problems, such as unsure sequence, single clone coverage, or low sequence quality annotated on the insert sequence records in GenBank, to the corresponding location in the current reference assembly.
- Available in GFF3 or ASN.1 format
- The files are found in organism-specific directories on the GRC’s public FTP site.
References
- 1.
- Schneider VA, Chen HC, Clausen C, Meric PA, Zhou Z, Bouk N, et al. Clone DB: an integrated NCBI resource for clone-associated data. Nucleic acids research. 2013;41(Database issue):D1070–8. [PMC free article: PMC3531087] [PubMed: 23193260]
- 2.
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, et al. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011;474(7351):337–42. [PMC free article: PMC3572410] [PubMed: 21677750]
- 3.
- Pettitt SJ, Liang Q, Rairdan XY, Moran JL, Prosser HM, Beier DR, et al. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nature methods. 2009;6(7):493–5. [PMC free article: PMC3555078] [PubMed: 19525957]
- 4.
- Skarnes WC, von Melchner H, Wurst W, Hicks G, Nord AS, Cox T, et al. A public gene trap resource for mouse functional genomics. Nature genetics. 2004;36(6):543–4. [PMC free article: PMC2716026] [PubMed: 15167922]
- 5.
- Hansen GM, Markesich DC, Burnett MB, Zhu Q, Dionne KM, Richter LJ, et al. Large-scale gene trapping in C57BL/6N mouse embryonic stem cells. Genome research. 2008;18(10):1670–9. [PMC free article: PMC2556270] [PubMed: 18799693]
- 6.
- Kidd JM, Cooper GM, Donahue WF, Hayden HS, Sampas N, Graves T, et al. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453(7191):56–64. [PMC free article: PMC2424287] [PubMed: 18451855]
- 7.
- Ventura M, Catacchio CR, Alkan C, Marques-Bonet T, Sajjadian S, Graves TA, et al. Gorilla genome structural variation reveals evolutionary parallelisms with chimpanzee. Genome research. 2011;21(10):1640–9. [PMC free article: PMC3202281] [PubMed: 21685127]
- 8.
- Safár J, Bartos J, Janda J, Bellec A, Kubaláková M, Valárik M, et al. Dissecting large and complex genomes: flow sorting and BAC cloning of individual chromosomes from bread wheat. The Plant journal. 2004Sep 39(6):960–8. [PubMed: 15341637]
- 9.
- Babiychuk E, Fuangthong M, Van Montagu M, Inze D, Kushnir S. Efficient gene tagging in Arabidopsis thaliana using a gene trap approach. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(23):12722–7. [PMC free article: PMC25099] [PubMed: 9356517]
- 10.
- Hsing YI, Chern CG, Fan MJ, Lu PC, Chen KT, Lo SF, et al. A rice gene activation/knockout mutant resource for high throughput functional genomics. Plant molecular biology. 2007;63(3):351–64. [PubMed: 17120135]
- 11.
- Lukacsovich T, Yamamoto D. Trap a gene and find out its function: toward functional genomics in Drosophila. Journal of neurogenetics. 2001;15(3-4):147–68. [PubMed: 12092900]
- 12.
- Cheung VG, Nowak N, Jang W, Kirsch IR, Zhao S, Chen XN, et al. Integration of cytogenetic landmarks into the draft sequence of the human genome. Nature. 2001;409(6822):953–8. [PMC free article: PMC7845515] [PubMed: 11237021]
- 13.
- Jang W, Yonescu R, Knutsen T, Brown T, Reppert T, Sirotkin K, et al. Linking the human cytogenetic map with nucleotide sequence: the CCAP clone set. Cancer genetics and cytogenetics. 2006;168(2):89–97. [PubMed: 16843097]
Publication Details
Author Information and Affiliations
Authors
Valerie Schneider, Ph.D.1.Affiliations
Publication History
Created: November 14, 2013.
Copyright
Publisher
National Center for Biotechnology Information (US), Bethesda (MD)
NLM Citation
Schneider V. Clone. 2013 Nov 14. In: The NCBI Handbook [Internet]. 2nd edition. Bethesda (MD): National Center for Biotechnology Information (US); 2013-.
![Figure 5. . Screenshot of results returned from an Entrez query of Clone DB (("gene targeting"[Library Type]) AND mouse[Organism]).](/books/NBK153524/bin/Clone-Image005.jpg)